Химия и химические
технологии/2.Теоретическая
химия
К.х.н. Бояркина О.В.
Мордовский государственный университет им. Н. П.
Огарева, Россия
Квантовофизические
методы исследования
соединений
бора
В настоящей работе проведено изучение
геометрического строения малых соединений бора: триметилборана,
триметоксиборана, тривинилборана и триаллилборана (см. рис. 1) методом
Хартри-Фока (HF) [1], методом функционала плотности (DFT) [2] и в рамках
теории возмущения Меллера-Плессета 2-го порядка (MP2) [3].
Ри
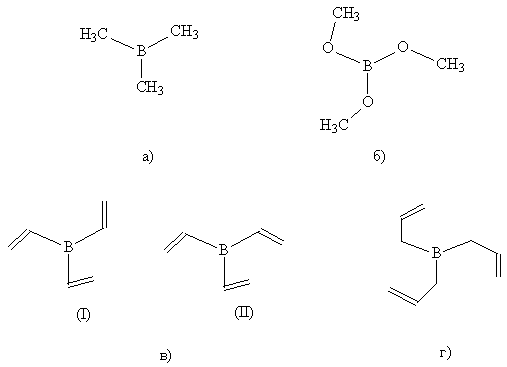
с. 1. Структура соединений бора: а) триметилборана, б) триметоксиборана,
в)
тривинилборана, г) триаллилборана
В соединениях триметилборана, триметоксиборана, тривинилборана и триаллилборана
бор трехкоординирован, поэтому наряду с другими подобными соединениями [4] в рассматриваемых молекулах имеются планарные
фрагменты BR3 (R=C, O). Исследования
[5] структуры
триметилборана (рис. 1 а)) методом дифракции электронов показали, что
триметилборан имеет плоский скелет с длиной связи d(B‒C)=1,578Å. Согласно исследованиям триметоксиборана [6] таким же методом планарный фрагмент B(OC)3 имеет точечную группу симметрии C3h, d(B‒O)=1,367(4)Å,
d(O‒C)=1,424(5)Å, α(BOC)=121,4°.
Согласно [7] известны 2 устойчивые конформации для тривинилборана I и II (рис. 1в)) с небольшой разницей в значениях полной энергии,
равной 0,3 ккал/моль: конформация I обладает плоской структурой с точечной группой
симметрии С3, конформация II —
менее плоская, структура характеризуется точечной группой симметрии С1.
Согласно данным метода электронографии [8] тривинилборан
имеет плоскую структуру с длинами связи
d(B‒C)=1,558Å и d(C=C)=1,370Å. При интерпретации структурных данных тривинилборана
необходимо использовать представления о делокализации π‒электронов на
вакантную p‒орбиталь атома бора. Данный
факт вызывает уменьшение длины связи B‒C и увеличение
длины связи С=C в B(CH=CH2)3
по сравнению с B(CH3)3 (1,578Å), B(CH2‒CH=CH2)3
(1,580Å) и H2C=CH2 (1,337Å), CH3‒CH=CH2 (1,342Å) соотвественно.
Исследования геометрического строения
молекулы триаллилборана (см. рис. 1г)))
методом газовой хроматографии [9] показали, что
молекула имеет симметрию С3. Значения длины связи составляют d(B‒C)=1,580Å, d(C‒C)=1,500Å, d(C=C)=1,330Å, значение угла α(CBC)=117,9°.
Оптимизация геометрии молекул
триметилборана, триметоксиборана, тривинилборана и триаллилборана
осуществлялась с помощью стандартного пакета прикладных программ GAMESS [10]. При
проведении расчетов были использованы 50 различных неэмпирических схем, которые
представлены в табл. 1. Используемые неэмпирические схемы расчета включают минимальные базисные наборы Поупла STO-nG [11, 12] (схемы расчета
4, 5, 18, 19, 27, 28, 36, 37, 45, 46), валентно-расщепленные
базисные наборы Поупла (SV) m-npG [13,
14] и Dunning/Hay (DH) [15] (схемы расчета 6-14, 20-23, 29-32, 38-41, 47-50 и 1,
15, 24, 33, 42 соответственно), а также трехэкспонентный валентный базисный
набор TZV
[16] (схемы расчета 2, 3, 16, 17, 25, 26, 34, 35, 43, 44).
Некоторые базисные наборы (3, 7, 9, 11, 17, 21, 23, 26, 30, 32, 35, 39, 41, 44,
48, 50) учитывают диффузионные (+) и поляризационные d- (*) и p-функции (**).
При проведении расчетов методом DFT были использованы обменные, корреляционные и гибридные
функционалы с различными поправками: PW91 [17] (схемы расчета 15-23), PBE [18] (схемы расчета
24-32), BLYP
[19] (схемы расчета 33-41) и B3LYP [20] (схемы расчета 42-50).
Таблица 1
Используемые неэмпирические схемы расчета
|
№ п/п |
Неэмпирические схемы |
№ п/п |
Неэмпирические схемы |
№ п/п |
Неэмпирические схемы |
|
1 |
HF/DH |
18 |
PW91/STO-3G |
35 |
BLYP/TZV** |
|
2 |
HF/TZV |
19 |
PW91/STO-6G |
36 |
BLYP/STO-3G |
|
3 |
HF/TZV** |
20 |
PW91/3-21G |
37 |
BLYP/STO-6G |
|
4 |
HF/STO-3G |
21 |
PW91/3-21+G** |
38 |
BLYP/3-21G |
|
5 |
HF/STO-6G |
22 |
PW91/6-31G |
39 |
BLYP/3-21+G** |
|
6 |
HF/3-21G |
23 |
PW91/6-31+G** |
40 |
BLYP/6-31G |
|
7 |
HF/3-21+G** |
24 |
PBE/DH |
41 |
BLYP/6-31+G** |
|
8 |
HF/6-21G |
25 |
PBE/TZV |
42 |
B3LYP/DH |
|
9 |
HF/6-21+G** |
26 |
PBE/TZV** |
43 |
B3LYP/TZV |
|
10 |
HF/6-31G |
27 |
PBE/STO-3G |
44 |
B3LYP/TZV** |
|
11 |
HF/6-31+G** |
28 |
PBE/STO-6G |
45 |
B3LYP/STO-3G |
|
12 |
MP2/3-21G |
29 |
PBE/3-21G |
46 |
B3LYP/STO-6G |
|
13 |
MP2/6-21G |
30 |
PBE/3-21+G** |
47 |
B3LYP/3-21G |
|
14 |
MP2/6-31G |
31 |
PBE/6-31G |
48 |
B3LYP/3-21+G** |
|
15 |
PW91/DH |
32 |
PBE/6-31+G** |
49 |
B3LYP/6-31G |
|
16 |
PW91/TZV |
33 |
BLYP/DH |
50 |
B3LYP/6-31+G** |
|
17 |
PW91/TZV** |
34 |
BLYP/TZV |
При изучении геометрического строения
рассматриваемых соединений бора проводилось сравнение рассчитанных значений
длины связи B-C, B-O, O-C, C-C, C=C при использовании различных неэмпирических схем
расчета с экспериментальными данными.
Для молекулы триметилборана использование
неэмпирических схем расчета 5, 16 и 43 приводит к совпадению длины связи d(B-C)расч. с d(B-C)эксп. Отметим, что в двух последних схемах расчета был использован метод DFT c трехэкспонентным валентным базисным набором TZV. Близкие значения рассчитанной длины связи B-C c экспериментальным значением показало использование
схем расчета 17, 19, 23, 25, 26, 28, 44, 49 и 50. Величина Δ1=|d(B-C)эксп. - d(B-C)расч.| для
них не превышает 0,002Å. Наибольшее отклонение от экспериментального
значения рассчитанной длины связи B-C показывает
использование неэмпирической схемы 13.
Результаты расчетов молекулы
триметоксиборана при использовании неэмпирических схем расчета 1-50 показали,
что использование схем расчета 1, 4, 10, 44 и 50 приводит к наименьшему
отклонению d(B-O)расч. от экспериментального
значения. Для схемы расчета 50 величина Δ2=|d(B-O)эксп.- - d(B-O)расч.|
составляет 0,003 Å, для схем расчета 1, 4 и 44 — 0,002 Å, для схемы 10 — 0,001 Å. К наибольшему
значению Δ2 приводит использование 36 и 39
неэмпирических схем (0,040 Å). В случае длины связи O-C в молекуле триметоксиборана наименьшее значение
Δ3=|d(O-C)эксп.-d(O-C)расч.|
характерно для неэмпирических схем
расчета 2, 4, 5, 44 и 50 (0,001, 0,003, 0,001, 0,003 и 0,003 Å,
соответственно). Наибольшее значение Δ3 показывает использование неэмпирических схем расчета
21, 30, 36 и 39 (0,060, 0,061, 0,063 и 0,074 Å, соответственно).
Как показывает сравнение рассчитанных
значений d(B-O) и d(O-C) c экспериментальными данными в молекуле
триметоксиборана, наиболее приемлемыми неэмпирическими схемами расчета для
данной молекулы с точки зрения соответствия экспериментальным данным являются
схемы 4, 44 и 50, а также 2 (Δ2=0,004 Å) и 10 (Δ3=0,007 Å).
В случае молекулы тривинилборана
конформации II (рис. 1в) для
неэмпирических схем расчета 5 и 20 экспериментальное и рассчитанное значения
длины связи d(B-C) в молекуле тривинилборана
совпадают. Для схем расчета 16, 27, 45 и 47 значение Δ4=|d(B-С)эксп. - d(B-С)расч.|
составляет 0,002, 0,001, 0,003 и 0,001 Å, cоответственно. Использование неэмпирических схем
расчета 11, 13 и 14 приводит к наибольшему отклонению d(B-C)расч. от d(B-C)эксп.
Значение рассчитанной длины связи B-C превышает
экспериментальное значение для данных схем расчета на 0,015, 0,016 и 0,015 Å, соответственно.
Отметим, что почти все использованные
неэмпирические схемы расчета приводят к заниженному значению рассчитанной длины
связи С=С в молекуле тривинилборана по сравнению с экспериментальным значением.
Причем к самому минимальному значению длины связи d(C=C) по сравнению с экспериментальным значением (1,370
Å) приводит использование 5 неэмпирической схемы расчета, для которой d(C=C)расч.=1,316 Å. Таким образом, можно отметить,
что большинство использованных неэмпирических схем расчета не позволяют учесть
делокализацию π-электронов двойной связи С=С на вакантную p-орбиталь атома бора, которая приводит к увеличению
длины связи С=С по сравнению с данным параметром, например, в этилене или
пропене. Наименьшее отклонение d(C=C)расч. от d(C=C)эксп. имеют неэмпирические схемы расчета, включающие
валентно-расщепленный базисный набор Dunning/Hay, а именно 15, 24 и 33. Величина
Δ5=|d(С=С)эксп. - d(С=С)расч.| для данных неэмпирических схем расчета имеет
значение 0,003, 0,002 и 0,002 Å,
соответственно.
Наиболее приемлемыми с точки зрения
соответствия экспериментальным данным для d(B-C) и d(C=C) является
использование для молекулы тривинилборана неэмпирических схем расчета 15 и 24.
Значение Δ4 для данных неэмпирических схем
расчета составляет 0,007 и 0,008 Å, соответственно.
Для молекулы триаллилборана использование
всех неэмпирических схем расчета 1-50 приводит к завышенным значениям длины
связи B-C
по сравнению с экспериментальным
значением. Можно выделить три схемы расчета, имеющие наименьшее отклонение d(B-C)расч. от d(B-C)эксп.
- 17, 26 и 44. Для данных схем расчета величина Δ6=|d(B-C)эксп. - d(B-C)расч.|
составляет 0,004, 0,006 и 0,005
Å, соответственно. Стоит отметить, что в данных неэмпирических
схемах расчета используется трехэкспонентный валентный базисный набор с
включением диффузных и поляризационных d- и p-функций.
Можно выделить также неэмпирические схемы
расчета, имеющие наибольшее значение Δ6: 13, 33, 36 и 37 (0,022, 0,021,
0,023 и 0,021 Å, соответственно.)
В случае длины связи d(C-C) в молекуле триаллилборана использование неэмпирических
схем расчета 23 и 26 приводит к совпадению данного параметра с
экспериментальным значением (1,500 Å), для схем расчета 32 и 44 величина Δ7=|d(С-C)эксп. - d(С-C)расч.| не
превыщает 0,001 Å. Расчеты молекулы триаллилборана в рамках
неэмпирических схем расчета 36 и 37 приводят к наибольшим значениям Δ7: 0,044 и 0,041
Å, соответственно.
Для длины связи d(C=C) в молекуле триаллилборана использование неэмпирической
схемы расчета 44 приводит к наилучшему приближению к экспериментальному
значению (1,330 Å). Значение Δ8=|d(C=C)эксп. - d(C=C)расч.| для
молекулы триаллилборана при использовании неэмпирической схемы расчета 44
составляет 0,002 Å.
Неэмпирические схемы 4, 5, 15 и 24
имеют наибольшее значение Δ8 для молекулы триаллилборана — 0,021,
0,022, 0,030 и 0,030 Å, соответственно. Таким образом, сравнение
результатов расчета длин связи B-C, C-C и С=С в
молекуле триаллилборана при использовании неэмпирических схем расчета 1-50
позволяет выделить наиболее приемлемую с точки зрения соответствия
экспериментальным данным неэмпирическую схему расчета 44 (Δ8=0,002 Å).
Полученные выводы, касающиеся выбора
наиболее приемлемой неэмпирической схемы расчета, из рассматриваемых 1-50 схем
расчета, для молекул триметилборана, триметоксиборана, тривинилборана и
триаллилборана с точки зрения соответствия экспериментальным данным, могут быть
полезны при проведении теоретических исследований в рамках различных
квантовофизических методов более протяженных неполимерных и полимерных
соединений бора. Результаты расчетов также могут быть полезны при проведении
теоретических исследований соединений бора, для изучения геометрического
строения и свойств которых используются представления о делокализации π-электронов ненасыщенной системы молекулы на вакантную p-орбиталь атома бора.
Литература:
- Roothaan C.C.J. New Developments in Molecular
Orbital Theory // Rev.Mod.Phys., 1951, v. 23. P. 69-89.
- Moeller C., Plesset M. S. Note on an
approximation treatment for many-electron systems // Phys. Rev., 1934, v.
46. P. 618-622.
- Kohn W., Sham L.J. Self-Consistent
Equations Including Exchange and Correlation Effects // Phys. Rev. 1965.
V.140 A. Р.1133.
- Михайлов, Б. М. Борорганические соединения в органическом синтезе / Б. М. Михайлов, Ю. Н. Бубнов. М., Наука, 1977,
516 с.
- Bartell L. S., Carroll B. L.
Electron‐Diffraction Study of the Structure of B(CH3)3
// J. Chem. Phys., 1965, v. 42, №
9. P.
3076‒3078.
- Steinberg, H. Organoboron Chemistry / H.
Steinberg. Wiley‒Interscience, New York, 1964, V. 1.; Grete
Gundersen. Molecular structure of gaseous methyl borate, B(OCH3)3
// J. Mol. Struct., 1976, v.
33, № 1, P. 79‒89.
- Odom J. D., Hall L.
W., Riethmiller S., Durig J. R.
Vibrational spectra and structure of trivinylborane // Inorg. Chem., 1974,
v. 13, № 1, P. 170–174; Durig J. R.,
Kim Young Hae, Little T.S..
Conformational stability of trivinylborane from vibrational spectra and ab
initio calculations // Spectrochimica Acta Part A: Molecular Spectroscopy,
1994, v. 50, № 3. P.
609–619.
- Foord A., Beagley В., Reade
W., Steer I. A. A gas‒phase electron‒diffraction study of
trvinylborane // J. Mol. Struct., 1975, v. 24., № 1. P. 131‒137.
- Вишневский, Ю. В. Исследование геометрического строения молекулы
триаллиборана методом газовой хроматографии / Ю. В. Вишневский, Л. В. Вилков, А. Н. Рыков, Н. М.
Карасев, Ю. Н. Бубнов, М. Е. Гурский // Известия Академии наук, сер. Хим., 2005, №1, С. 98‒101.
- Schmidt M.W., Baldridge
K.K., Boatz J.A., Elbert S.T., Gordon M.S., Jensen J.H., Koseki S., Matsunaga
N., Nguyen K.A., Su S.J., Windus T.L., Dupuis
M., Montgomery J.A. General Atomic
and Molecular Electronic Structure System // J. Comput. Chem., 1993, v.
14. P. 1347-1363.
- Hehre W.J., Stewart
R.F., Pople J.A..
Self‐Consistent Molecular‐Orbital Methods. I. Use of Gaussian
Expansions of Slater‐Type Atomic Orbitals // J. Chem. Phys., 1969, v. 51. P. 2657-2664.
- Hehre W.J., Ditchfield R., Stewart R.F., Pople J.A. Self‐Consistent Molecular Orbital Methods.
IV. Use of Gaussian Expansions of Slater‐Type Orbitals. Extension to
Second‐Row Molecules // J. Chem. Phys, 1970, v. 52. P. 2769 -2773.
- Binkley J.S., Pople J.A., Hehre W.J. Self-consistent molecular orbital methods. 21.
Small split-valence basis sets for first-row elements // J. Am. Chem. Soc., 1980, v. 102. P. 939-947.
- Gordon M.S., Binkley J.S., Pople J.A., Pietro
W.J., Hehre W.J. Self-consistent
molecular-orbital methods. 22. Small split-valence basis sets for
second-row elements // J. Am. Chem. Soc., 1982,
v. 104.
P. 2797-2803.
- Dunning Jr. T.H., Hay P.J.
in `Modern Theoretical Chemistry', Vol. 3, ed. H.F. Schaefer, Plenum, New
York, 1977.
- Dunning Jr. T.H. Gaussian Basis Functions for Use in
Molecular Calculations. III. Contraction of (10s6p) Atomic Basis Sets for the
First‐Row Atoms // J. Chem. Phys., 1971, v. 55. P. 716-723.
- Perdew
J. P., Wang Y. Accurate and simple density functional for the electronic
exchange energy: Generalized gradient approximation // Phys. Rev. B. 1986. V.33. P. 8800-8802.
- Perdew J.P., K. Burke,
Ernzerhof M. Generalized Gradient Approximation Made Simple // Phys. Rev. Lett., 1996, Vol. 77. P. 3865-3868.
- Lee C., Yang W., Parr R.G.Development of the Colle-Salvetti
correlation-energy formula into a functional of the electron density //
Phys. Rev., 1988, B37. P. 785-789.
- Becke A.D. Density‐functional thermochemistry. III. The role
of exact exchange // J.Chem.Phys., 1993, v. 98. P. 5648-5642.