Химия и химические
технологии/2.Теоретическая
химия
К.х.н. Бояркина О.В.
Мордовский государственный университет им. Н. П.
Огарева, Россия
Применение
квантовофизических методов для исследования
боразола и бороксола
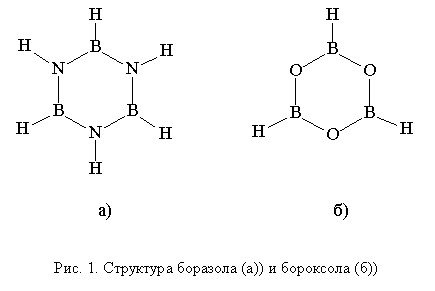
Боразол и бороксол (рис. 1) представляют
собой шестичленные плоские циклы, точечной группы симметрии D3h. Благодаря особенностям строения молекул и возможной
делокализации неподеленных пар атомов азота и кислорода на вакантную орбиталь
атома бора данные молекулы можно рассматривать как неорганические аналоги
бензола [1,
2].
Согласно электронографическим
исследованиям [3] все связи между атомами азота
и бора в боразоле являются равноценными, и их длина (1,436 Å),
имеет среднее значение между длиной простой связи B‒N (1,54 Å) и величиной
1,36 Å для двойной связи B=N, приближаясь
по величине к длине связи C=C в
бензоле (1,397 Å). Согласно исследованиям бороксола методом дифракции
электронов [4] значение длины связи d(B‒O)=1,3758±0,0021Å, значение валентного угла в кольце составляет 120±0,64°.
Производные бороксола и боразола находят
большое применение в качестве органических проводниковых материалов [5], материалов
обладающих оптическими нелинейными свойствами [6, 7] и др.
В настоящей работе изучение
геометрического строения боразола и бороксола проведено методом Хартри-Фока (HF) [8], методом
функционала плотности (DFT) [9] и в рамках теории возмущения Меллера-Плессета 2-го
порядка (MP2)
[10] при использовании 50 неэмпирических схем расчета,
представленных в табл. 1.
Таблица 1
Используемые неэмпирические схемы расчета
|
№ п/п |
Неэмпирические схемы |
№ п/п |
Неэмпирические схемы |
№ п/п |
Неэмпирические схемы |
|
1 |
HF/DH |
18 |
PW91/STO-3G |
35 |
BLYP/TZV** |
|
2 |
HF/TZV |
19 |
PW91/STO-6G |
36 |
BLYP/STO-3G |
|
3 |
HF/TZV** |
20 |
PW91/3-21G |
37 |
BLYP/STO-6G |
|
4 |
HF/STO-3G |
21 |
PW91/3-21+G** |
38 |
BLYP/3-21G |
|
5 |
HF/STO-6G |
22 |
PW91/6-31G |
39 |
BLYP/3-21+G** |
|
6 |
HF/3-21G |
23 |
PW91/6-31+G** |
40 |
BLYP/6-31G |
|
7 |
HF/3-21+G** |
24 |
PBE/DH |
41 |
BLYP/6-31+G** |
|
8 |
HF/6-21G |
25 |
PBE/TZV |
42 |
B3LYP/DH |
|
9 |
HF/6-21+G** |
26 |
PBE/TZV** |
43 |
B3LYP/TZV |
|
10 |
HF/6-31G |
27 |
PBE/STO-3G |
44 |
B3LYP/TZV** |
|
11 |
HF/6-31+G** |
28 |
PBE/STO-6G |
45 |
B3LYP/STO-3G |
|
12 |
MP2/3-21G |
29 |
PBE/3-21G |
46 |
B3LYP/STO-6G |
|
13 |
MP2/6-21G |
30 |
PBE/3-21+G** |
47 |
B3LYP/3-21G |
|
14 |
MP2/6-31G |
31 |
PBE/6-31G |
48 |
B3LYP/3-21+G** |
|
15 |
PW91/DH |
32 |
PBE/6-31+G** |
49 |
B3LYP/6-31G |
|
16 |
PW91/TZV |
33 |
BLYP/DH |
50 |
B3LYP/6-31+G** |
|
17 |
PW91/TZV** |
34 |
BLYP/TZV |
Оптимизация геометрии молекул боразола и
бороксола осуществлялась с помощью стандартного пакета прикладных программ GAMESS [11]. Используемые
неэмпирические схемы расчета включают
минимальные базисные наборы Поупла STO-nG [12,
132], валентно-расщепленные базисные наборы Поупла (SV) m-npG [14, 15] и Dunning/Hay (DH) [16], а также
трехэкспонентный валентный базисный набор TZV [17]. Некоторые
базисные наборы учитывают диффузионные (+) и поляризационные d- (*) и p-функции (**).
При проведении расчетов методом DFT были использованы обменные, корреляционные и гибридные
функционалы с различными поправками: PW91 [18], PBE [19], BLYP [20] и B3LYP [21].
В результате проводилось сравнение
рассчитанных значений длин связей d(B-N) и d(B-O) в молекулах боразола и бороксола c экспериментальными данными. На рис. 2 представлены
значения рассчитанной длины связи d(B-N) в
молекуле боразола в сравнении с экспериментальным значением.
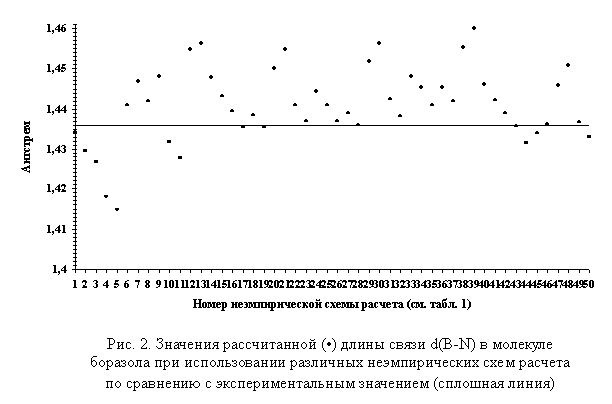
Как видно из рис. 2, использование неэмпирических
схем расчета 17, 19, 28, 43 и 46 приводит к совпадению рассчитанной длины связи
d(B-N)
в молекуле боразола с экспериментальным
значением. Для схем расчета 23, 26, 46 и 49 значение Δ1=|d(B-N)эксп. - d(B-N)расч.| не
превышает 0,0009 Å. Максимальное значение Δ1 (0,0241 Å) характерно для неэмпирической схемы
расчета 39.
На рис. 3 представлены значения
рассчитанной длины связи d(B-O) в
молекуле бороксола по сравнению с экспериментальным значением.
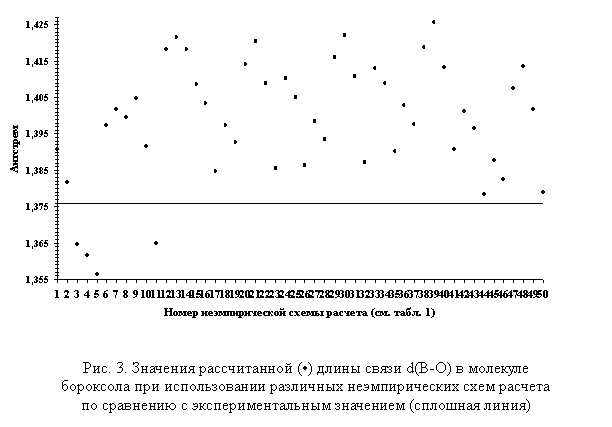
Как видно из рис. 3, минимальные значения Δ2=|d(B-O)эксп. - d(B-O)расч.| получено при использовании неэмпирических схем
расчета 44 и 50 (0,0026 и 0,0032 Å, соответственно), максимальное
значение — 39 (0,0498 Å).
Полученные результаты, представленные в
данной работе, могут быть использованы при теоретическом исследовании строения
и свойств более протяженных неполимерных и полимерных производных боразола и
бороксола.
Литература:
1. Fowler P. W., Steiner E. Ring
Currents and Aromaticity of Monocyclic π-Electron Systems C6H6,
B3N3H6, B3O3H3,
C3N3H3, C5H5-,
C7H7+, C3N3F3,
C6H3F3, and C6F6 // J. Phys. Chem. A, 1997, v. 101, №7. P. 1409–1413.
2. Engelberts J. J., Havenith R. W. A., van Lenthe J. H., Jenneskens L. W., Fowler P. W. The
Electronic Structure of Inorganic Benzenes: Valence Bond and Ring-Current
Descriptions // Inorg. Chem., 2005, v. 44. P. 5266-5272.
3. Stock A., Wiebe
R. Die Konstitution des B3N3H6
// Z. anorg. Allg. Chem., v. 203, №1, 1932. P. 228‒234.
4. Chang C.-H.,
Porter R. F., Bauer S. H.. Molecular structure of boroxin, H3B3O3,
determined by electron diffraction // Inorg. Chem., v. 8, № 8, 1969. PP. 1689–1693.
5. Qin Y., Cui C.,
Jӓkle F.
Silylated Initiators for the Efficient Preparation of Borane-End-Functionalized
Polymers via ATRP // Macromolecules, 2007,
v. 40, № 5. P. 1413‒1420.
6.
Katan C., Mongin O., Alcaraz G., Vaultier M., Boucekkineb A.,
Blanchard‒Desce M.. Improved Transparency-Nonlinearity Trade-Off with
Boroxine‒Based Octupolar Molecules // Linear and Nonlinear Optics of
Organic Materials IV, Denver: United States, 2004.
7.
Sastre A.,
Torres T., Díaz-García
M. A., Agulló-López
F., Dhenaut C., Brasselet S., Ledoux I.,
Zyss J. Subphthalocyanines: Novel Targets for
Remarkable Second‒Order Optical Nonlinearities // J. Am. Chem. Soc., v.
118, № 11, 1996. P. 2746–2747.
8.
Roothaan C.C.J. New Developments in Molecular Orbital Theory //
Rev.Mod.Phys., 1951, v. 23. P. 69-89.
9. Moeller C., Plesset
M. S. Note on an approximation treatment for many-electron systems // Phys.
Rev., 1934, v. 46. P. 618-622.
10. Kohn W., Sham L.J. Self-Consistent Equations Including Exchange and
Correlation Effects // Phys. Rev. 1965. V.140 A. Р.1133.
11. Schmidt
M.W., Baldridge K.K., Boatz J.A.,
Elbert S.T., Gordon M.S., Jensen
J.H., Koseki S., Matsunaga N., Nguyen K.A., Su
S.J., Windus T.L., Dupuis M.,
Montgomery J.A. General Atomic and Molecular Electronic Structure System
// J. Comput. Chem., 1993, v. 14. P. 1347-1363.
12.
Hehre W.J., Stewart
R.F., Pople J.A.. Self‐Consistent
Molecular‐Orbital Methods. I. Use of Gaussian Expansions of
Slater‐Type Atomic Orbitals // J. Chem. Phys., 1969, v. 51. P. 2657-2664.
13.
Hehre W.J., Ditchfield R., Stewart R.F., Pople J.A.
Self‐Consistent Molecular Orbital Methods. IV. Use of Gaussian Expansions
of Slater‐Type Orbitals. Extension to Second‐Row Molecules // J. Chem. Phys, 1970,
v.
52. P.
2769
-2773.
14.
Binkley J.S., Pople J.A.,
Hehre W.J. Self-consistent molecular orbital methods. 21. Small
split-valence basis sets for first-row elements // J. Am. Chem. Soc., 1980,
v. 102.
P. 939-947.
15.
Gordon M.S., Binkley J.S., Pople J.A., Pietro
W.J., Hehre W.J. Self-consistent
molecular-orbital methods. 22. Small split-valence basis sets for second-row
elements //
J. Am. Chem. Soc., 1982, v. 104. P. 2797-2803.
16.
Dunning Jr. T.H.,
Hay P.J. in `Modern Theoretical Chemistry', Vol. 3, ed. H.F. Schaefer, Plenum,
New York, 1977.
17.
Dunning Jr.
T.H. Gaussian Basis Functions for Use
in Molecular Calculations. III. Contraction of (10s6p) Atomic Basis Sets for the
First‐Row Atoms // J. Chem. Phys., 1971,
v.
55. P. 716-723.
18.
Perdew J. P., Wang Y. Accurate and simple density
functional for the electronic exchange energy: Generalized gradient
approximation // Phys. Rev. B. 1986. V.33. P. 8800-8802.
19. Perdew J.P., K. Burke, Ernzerhof M. Generalized Gradient
Approximation Made Simple // Phys. Rev. Lett., 1996,
Vol. 77. P. 3865-3868.
20.
Lee C., Yang W., Parr
R.G. Development of the Colle-Salvetti correlation-energy formula into a
functional of the electron density // Phys. Rev., 1988, B37. P. 785-789.
21.
Becke A.D.
Density‐functional thermochemistry. III. The role of exact exchange //
J.Chem.Phys., 1993, v. 98. P. 5648-5642.