Коровашкина
А.С., Квач С.В., Зинченко А.И.
Институт
микробиологии НАН Беларуси, 220141, Минск, ул. Купревича, 2
Получение термостабильной ДНК-полимеразы
с повышенной устойчивостью к цельной крови
При постановке полимеразной цепной реакции
(ПЦР) наиболее часто в качестве ДНК-полимеразы используется фермент из
термофильной бактерии Thermus aquaticus
(Taq-ДНК-полимераза). Однако Taq-ДНК-полимераза обладает рядом
существенных недостатков. Среди них: большое количество ошибок при синтезе ДНК;
высокая чувствительность к различным ингибиторам ПЦР; недостаточная
термостабильность; низкая процессивность; наличие 5'→3'-экзонуклеазной
активности.
В свое время была предложена KlenTaq-ДНК-полимераза (фрагмент Кленова) – «укороченная
версия» Taq-ДНК-полимеразы (отсутствуют
первые 279 аминокислотных остатков), лишенная 5'→3'-экзонуклеазной
активности. Такой фермент более термостабилен, более точен (хотя, менее
процессивен), чем Taq-ДНК-полимераза.
Важной особенностью KlenTaq-ДНК-полимеразы
является ее устойчивость к цельной крови (до 5–10%), в то время как нативный
фермент ингибируется уже при наличии в реакционной смеси 0,1–1% крови [1].
Для преодоления некоторых из перечисленных
выше недостатков ДНК-полимераз в литературе был предложен подход, основанный на
получении химерных белков, включающих кроме полимеразного домена
неспецифический ДНК-связывающий домен, повышающий сродство полимеразы к матрице
и, как следствие, повышающий процессивность, скорость синтеза, точность
функционирования, устойчивость к высокой ионной силе раствора [2–4].
В связи с вышеизложенным, целью настоящей работы явилось получение «химерной» ДНК-полимеразы с улучшенными свойствами за счет
присоедине-ния к каталитическому домену KlenTaq-ДНК-полимеразы
неспецифического ДНК-связывающего белка (Sso7d) бактерии Sulfolobus solfataricus.
Материалы
и методы исследования. Участок гена KlenTaq-ДНК-плимеразы
(KT), начинающийся с 280-го аминокислотного остатка, был выделен методом ПЦР с
использованием в качестве матрицы геномной ДНК T. aquaticus. Для амплификации KT использовали следующую пару
праймеров: F
(5'-GAATTCCTCCTCCACGAGTTCGGCCTTC-3')
и R
(5'-TATGTCGACTTAGTGATGGTGATGGTGATGCTCCTTGGCGGAGAGCCAGT-3').
В 5'-окончания праймеров были встроены сайты рестрикции (выделены курсивом)
эндонуклеаз EcoRI (F) и SalI (R). В R-праймер также была
введена последовательность, кодирующая полигистидиновый олигопептид (жирный
шрифт). Синтезированный с помощью ПЦР продукт обрабатывали рестрикционными
эндонуклеазами EcoRI и SalI
и клонировали в векторе pET22b+ (Novagen, США), предварительно обработанный
этими же рестриктазами. В результате была получена конструкция pET22KT.
Нуклеотидная
последовательность, кодирующая белок Sso7d
(ген sso7d), была
получена из базы данных GenBank (№ 1453539). Оптимизацию кодонов для экспрессии
белка в E. coli проводили в программе
Gene Designer 2.0 (DNA 2.0, США). Дополнительно с 5'-конца гена вводили сайт
узнавания рестрикционной эндонуклеазы NdeI,
а с 3'-конца – последовательность, кодирующую линкерный олигопептид и сайт
узнавания рестрикционной эндонуклеазы EcoRI.
Дизайн олигонуклеотидов для сборки гена, кодирующего белок Sso7d, проводили с
использованием программы TmPrime version 3.0 (http://prime.ibn.a-star.edu.sg/).
Сборку нуклеотидной последовательности sso7d проводили с помощью ПЦР [5].
Синтезированную
с помощью ПЦР нуклеотидную последовательность sso7d обрабатывали смесью рестриктаз NdeI и EcoRI и встраивали
в полученный ранее и предварительно обработанный этими же рестриктазами вектор
pET22KT. Полученной плазмидой (pET22KT-Ssо7d) трансформировали
штамм E. coli BL21(DE3). Клетки-трансформанты выращивали на LB-среде до оптической плотности 0,6
(λ = 600 нм), затем проводили индукцию синтеза белка 1 мМ ИПТГ и
продолжали культивирование в течение 5 ч. По окончании выращивания клетки осаждали центрифугированием, ресуспендировали
в буфере, содержащем 50 мМ NaH2PO4, 300 мМ NaCl и 20 мМ
имидазол (рН 8,0). После ультразвуковой дезинтеграции
клеток лизат прогревали 30 мин при 70оС, осветляли центрифугированием
и супернатант наносили на хроматографическую колонку со смолой Ni-NTA (Qiagen,
США). Выделение белка проводили согласно инструкции фирмы-производителя.
Полученный раствор фермента диализовали против 1000-кратного объема буфера (10
мМ Трис-HC, рН 8,0 + 100 мМ KCl + 1 мМ ЭДТА + 2 мМ β-меркаптоэтанол + 0,5% Твин-20). Полученный после диализа препарат
белка разводили в 3 раза 10 мМ Трис-HCl-буфером (рН 8,0), содержащим 100 мМ
KCl, 1 мМ ЭДТА, 2 мМ β-меркаптоэтанол, 0,5% Твин-20 и 50% глицерин.
Определение активности Sso7d-KlenTaq-ДНК-полимеразы проводили с помощью ПЦР. Реакционная смесь (30
мкл) состояла из: синтетических олигонуклеотидов (F 5'-GTCTACCAGGCATTCGCTTCAT-3' и R 5'-CTGTGAATGCTGCGACTACGAT-3') (по 5 пмоль каждого),
четырех природных дезоксинуклеозидтрифосфатов (каждый в концентрации 0,2 мМ), 5
нг плазмидной ДНК, MgCl2 (3 мМ), KCl (90 мМ), Трис-HCl, pH 8,8 (50 мМ) и
0,05% Твина-20. В реакционную смесь вносили по 1 мкл двукратных разведений
очищенной Sso7d-KlenTaq-ДНК-полимеразы. Программа
амплификации: 2 мин 95оС, (15 с 95оС; 15 с 65оС;
30 с 72оС) – 30 циклов. Затем 3 мкл ПЦР-смеси вносили в 200 мкл
10-кратного интеркалирующего красителя Sybre green (Sigma, США), разведенного в 1660 раз ТЕ-буфером, и измеряли
флуоресценцию с помощью прибора Qubit
fluoro-meter (Invitrogen, США). За 1 ед. активности Sso7d-KlenTaq-ДНК-полимеразы
принимали такое ее количество, которое по флуоресценции соответствовало 1 ед.
коммерческого препарата Taq-ДНК-полимеразы
(Sileks, Россия).
Результаты
исследования. Из литературных
источников известно, что Sso7d-домен
позволяет значительно улучшить сродство ДНК-полимеразы к матрице и, как
следствие, повысить процессивность, скорость синтеза, устойчи-вость к высокой
ионной силе и различным ингибиторам [4]. Основываясь на этих данных, мы
предположили, что полученная химерная ДНК-полимераза будет также более
устойчива к ингибированию цельной кровью.
Для определения устойчивости Sso7d-KlenTaq-ДНК-полимеразы к цельной крови мы провели проверку наличия
точечной мутации в гене, кодирующем фактор
свёртываемости крови V (фактор Лейдена). Как известно,
мутация G1691→A в этом гене приводит к повышению риска развития
тромбоэмболии вен у гетерозигот в 8 раз, у гомозигот – в 80–100 раз [6]. Хотя
только 5% здорового населения являются носителями мутантного аллеля,
рассматриваемая мутация присутствует у 40% пациентов с диагнозом тромбоз вен.
Таким образом, своевременное выявление данной мутации необходимо для правильной
постановки диагноза и назначения адекватного лечения.
Для проверки влияния
ЭДТА-стабилизированной крови на ПЦР, проводимой при помощи полученной нами Sso7d-KlenTaq-ДНК-полимеразы, кровь здоровых доноров вносили в различных
концентрациях (1–20%, v/v) в реакционную смесь. Для амплификации участка гена,
кодирующего фактор Лейдена [6], использовали синтетические олигонуклеотиды (F
5'-TGCCCAGTGCTTAACAAGACCA-3' и R 5'-TGTTATCACACTGGTGCTAA-3').
Из электрофореграммы (рис. 1) видно, что
полученный нами ампликон имеет размер приблизительно равный 267 п.о., что
соответствует теоретически-рассчитанному. Таким образом, можно сделать вывод,
что Sso7d-KlenTaq-ДНК-полимераза проявляет высокую
ферментативную активность даже при повышенных концентрациях цельной крови (до
20%).
Наличие мутации определяли при помощи
обработки полученных при амплификации проб с применением эндонуклеазы
рестрикции MnlI. Известно, что при
рестрикции дикого аллеля образуются продукты длиной 67, 37 и 163 п.о., в то
время как при рестрикции мутантного аллеля размеры получаемых фрагментов
составляют 67 и 200 п.о. Анализ полученных фрагментов проводили с помощью
агарозного гель-электрофореза (рис. 2).
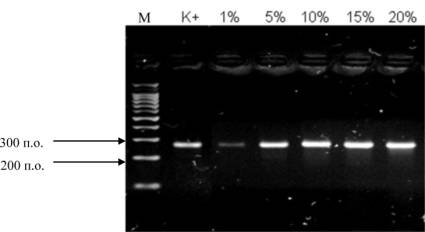
Рис. 1 – Электрофореграмма амплифицированного
сегмента (содержащего нуклеотид № 1691) гена, кодирующего фактор Лейдена.
M - фрагменты ДНК c известным количеством пар оснований;
K+ -
амплификация участка гена с использованием 10 нг очищенной ДНК;
1–20% - концентрация цельной крови в образце.
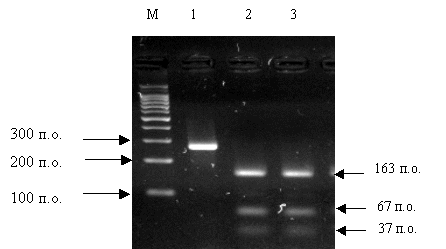
Рис. 2 – Электрофореграмма продуктов рестрикции
сегмента (содержащего нуклеотид № 1691) гена, кодирующего фактор Лейдена.
M - фрагменты ДНК c
известным количеством пар оснований; 1 – нативный сегмент; 2–3 - фрагменты
полученные в результате обработки сегмента рестриктазой MnlI.
Из электрофореграммы на рис. 2 видно, что
в обоих образцах содержатся фрагменты ДНК длиной 37, 67 и 163 п.о., что
соответствует дикому аллелю. Следовательно, данные пациенты гомозиготны по
дикому аллелю, т.е. не несут мутацию фактора Лейдена.
Таким образом, показано, что полученный
нами химерный фермент (Sso7d-KlenTaq-ДНК-полимераза) выдерживает высокие концентрации цельной крови
(до 20%) и может быть использован в качестве одного из компонентов
аналитических тест-систем в клинико-лабораторных исследованиях.
Литература:
1. Kermekchiev M.B., Kirilova L.I., Vail E.E., Barnes W.M. Mutants of Taq DNA polymerase resistant to PCR inhibitors allow DNA amplification from whole blood and crude soil samples // Nucleic Acids Res. 2009. – Vol. 37, № 5. – e40.
2. Pavlov A.R.,
Belova G.I., Kozyavkin S.A., Slesarev A.I. Helix-hairpin-helix motifs confer salt resistance and processivity on chimeric DNA polymerases
/ A.R. Pavlov [et al] // Proc. Natl Acad. Sci. USA. – 2002. – Vol. 99. – P. 13510–13515.
3. Wang Y., Prosen D.E., Mei L., Sullivan J.C., Finney M., Vander Horn P.B. A novel strategy to engineer DNA polymerases for enhanced processivity and improved performance in vitro //Nucleic
Acids Res. –
2004. – Vol. 32, № 3. – P.
1197–1207.
4. Sun S., Geng L., Shamoo Y. Structure and enzymatic properties of a
chimeric bacteriophage RB69 DNA polymerase and single-stranded DNA binding
protein with increased processivity // Proteins. – 2006. – Vol. 65, № 1. – P.
231–238.
5. Wu G., Wolf J.B., Ibrahim A.F., Vadasz S., Gunasinghe M., Freeland
S.J. Simplified gene synthesis: a one-step approach to PCR-based gene
construction // J. Biotechnol. 2006. Vol. 124, № 3. Р. 496–503.
6. Bertina R.M., Koeleman B.P., Koster T., Rosendaal F.R., Dirven R.J.,
de Ronde H., van der Velden P.A., Reitsma
P.H. Mutation in blood coagulation
factor V associated with resistance to activated protein C // Nature.
– 1994. – Vol. 369, № 6475. –
P. 64–77.