Рыбин Т.В., д.х.н. Белик
А.В.
Челябинский
государственный университет, Россия.
Квантовохимическое исследование
силовых полей соединений, участвующих в перегруппировке
Боултона – Катрицкого
Согласно
работам [1-2], производные
нитробензофуроксанов представляют огромный практический и теоретический
интерес.
Настоящее исследование посвящено
рассмотрению особенностей силовых элементов матрицы F [3-5], вычисленной в координатах ![]() [6,7], ряда
соединений класса бензофуроксанов, участвующих в прямой перегруппировке
Боултона-Катрицкого (БК) [2]. Начальные и конечные соединения перегруппировки представлены в таблице 1.
[6,7], ряда
соединений класса бензофуроксанов, участвующих в прямой перегруппировке
Боултона-Катрицкого (БК) [2]. Начальные и конечные соединения перегруппировки представлены в таблице 1.
Расчеты выполнены в рамках теории
функционала плотности [8] методом DFT B3LYP/6-31G [9,10]. В равновесной геометрии было вычислено силовое поле
в декартовой системе координат (Fx), которое затем было переведено в
координаты ![]() согласно процедуре,
описанной в работах [4,5,11].
В общем случае результат расчета зависит от выбора набора «векторов связей»
и ориентации «собственной системы координат» каждого из этих «векторов связей». Для удобства химической интерпретации результатов,
вектора связей совмещают с химическими связями в соединениях. Чтобы набор
координат был независимым, необходимо взять количество «векторов связей» на единицу меньше, чем
число атомов в молекуле. Тогда размерность матрицы
согласно процедуре,
описанной в работах [4,5,11].
В общем случае результат расчета зависит от выбора набора «векторов связей»
и ориентации «собственной системы координат» каждого из этих «векторов связей». Для удобства химической интерпретации результатов,
вектора связей совмещают с химическими связями в соединениях. Чтобы набор
координат был независимым, необходимо взять количество «векторов связей» на единицу меньше, чем
число атомов в молекуле. Тогда размерность матрицы ![]() будет (3N – 3)×(3N – 3), где N – количество атомов в молекуле. Как показано в [12], для характеристики «жесткости связи»
удобно использовать величину, представляющую след субматрицы (3×3) каждого из «векторов связей». Такие величины названы
обобщенными значениями силовых коэффициентов.
будет (3N – 3)×(3N – 3), где N – количество атомов в молекуле. Как показано в [12], для характеристики «жесткости связи»
удобно использовать величину, представляющую след субматрицы (3×3) каждого из «векторов связей». Такие величины названы
обобщенными значениями силовых коэффициентов.
В настоящей работе в качестве «векторов связей» выбраны такие связи, которые наиболее
«полно» участвуют в исследуемой перегруппировке (выделены на рисунке 1). Полученные
расчетные данные обобщенных значений силовых коэффициентов приведены в таблице 2.
Таблица 1
Соединения,
участвующие в прямой перегруппировке БК
|
№ |
Реакция |
|
1-2 |
|
|
3-4 |
|
|
5-6 |
|
|
7-8 |
|
|
9-10 |
|
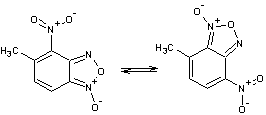
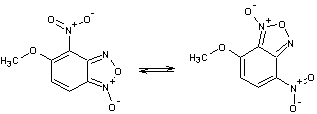
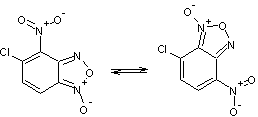
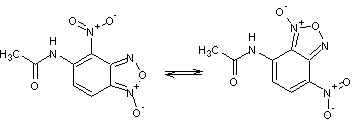
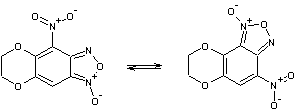
Очевидно, что в процессе
перегруппировки происходит перераспределение связей в молекуле. Поэтому важно
правильно выбрать пары связей для их сравнения. Так в случае прямой перегруппировки 1-2, связь N10–О11 следует рассматривать в паре со связью N1–O2.
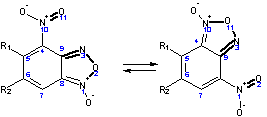
Рис. 1. Схема перегруппировки БК с выделенными
«векторами связей», которые рассмотрены в таблице 2
Таблица 2
Расчетные
значения обобщенных силовых коэффициентов некоторых связей (mdyn/Å) для исследованных соединений
|
№ |
(N–O) |
(C=N) |
(N=O) |
(N(O)–O) |
|
1 |
16.635 |
20.467 |
10.791 |
12.672 |
|
2 |
16.712 |
20.476 |
10.787 |
12.648 |
|
3 |
16.688 |
20.463 |
10.595 |
12.724 |
|
4 |
16.831 |
20.601 |
10.718 |
12.462 |
|
5 |
23.591 |
25.987 |
10.829 |
21.074 |
|
6 |
24.916 |
28.003 |
10.852 |
22.925 |
|
7 |
22.995 |
25.923 |
10.888 |
21.367 |
|
8 |
24.296 |
27.160 |
10.653 |
22.946 |
|
9 |
30.211 |
32.773 |
10.620 |
27.979 |
|
10 |
31.825 |
34.395 |
8.924 |
29.262 |
Жирным шрифтом в таблице отмечены
наибольшие значения обобщенных силовых
коэффициентов связей из полученных для выбранной пары соединений.
Анализируя полученные данные можно
отметить, что для большинства случаев в конечных соединениях перегруппировки БК связь N–O
(рвущаяся в ходе перегруппировки и снова образующаяся) становится «прочнее» по сравнению с аналогичной в
исходных соединениях. Такая же ситуация наблюдается и для
связи С=N.
Таким образом, можно заключить, что в
процессе перегруппировки образуется новая связь атома N3 с нитрогруппой, которая получается прочнее, чем в
исходном соединении, а так же прочнее становится связь этого атома с бензольным кольцом.
Литература:
1.
Хмельницкий
Л.И., Новиков С.С., Годовикова Т.И. Химия фуроксанов (Строение и синтез). М.: Наука. 1996. 383с.
2.
Хмельницкий
Л.И., Новиков С.С., Годовикова Т.И.Химия фуроксанов (Реакции и применение). М.: Наука.
1996. 430с.
3.
Волькенштейн
М.В., Грибов Л.А., Ельяшевич М.А., Степанов Б.И. Колебания молекул. М.: Наука. 1972. 699с.
4.
Белик
А.В. Теория и практика расчета колебаний молекул. Челябинск: Изд-во БашГУ. 1985. 47с.
5.
Белик
А.В., Шляпочников В.А. Квантовохимическая оценка силового поля аммиака в
координатах Хδ0 // Изв. АН СССР. Сер.хим. 1985. №3.
С.697-699.
6.
Маянц
Л.С., Шалтупер Г.Б. Новый подход к полному расчету колебаний любых молекул. // Докл.АН
СССР. 1972. №206. С.657-660.
7.
Mayants L.S., Shaltuper G.B. General
methods of analyzing molecular vibrations. Journal
of Molecular Structure. 1975. Vol.24. P. 409-431.
8. Кон В. Электронная
структура вещества – волновые функции и функционалы плотности. Успехи физ. наук (Нобелевские лекции по
химии – 1998). 2002. Т.172. №3. С.336-348.
9. Becke A.D. Density-functional thermochemistry. III. The role of exact
exchange. J. Chem. Phys. 1993. Vol.98.
P.5648-5652.
10. Stephens
P.J., Devlin F., Chabalowski C.F., Frisch M.J. Ab Initio Calculation of Vibrational Absorption and Circular
Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994. Vol.98. P.11623-11627.
11. Савчик Д.В., Балыкин В.П.,
Белик А.В. Решение колебательной задачи при использовании ![]() координат на примере молекулы аммиака // Вестник
Челябинского госуниверситета. Физика. 2010.№12. Вып.7. С. 73-77.
координат на примере молекулы аммиака // Вестник
Челябинского госуниверситета. Физика. 2010.№12. Вып.7. С. 73-77.
12.
Белик А.В., Федотова Е.И. Квантово-химическое
исследование силового поля нитрометана в координатах ![]() // Бутлеровские сообщения.
2011. Т.25. №5. с. 60-63.
// Бутлеровские сообщения.
2011. Т.25. №5. с. 60-63.