Химия и
химические технологии/2. Теоретическая химия
Romanova K.A.,
Galyametdinov Yu.G.
Kazan
National Research Technological University, Russia
Theoretical simulation of lanthanide(III) complexes
promising for luminescent functional materials
Lanthanide (Ln) coordination
compounds have a great potential as luminescent materials due to their remarkable magnetic and
optical properties especially for
optoelectronic devices, flat and flexible displays, organic light emitting
diodes, luminescent biological probes and solar cells. Liquid-crystalline Ln(III)
complexes can form optically transparent films with polarized luminescence,
whose intensity can be controlled by magnetic and electric fields, temperature,
and laser irradiation.
Photophysical properties of Ln(III) complexes are mainly defined by their ligand
environment [1]. Organic ligands in these molecules provide the transfer of the
excitation energy onto the emissive Ln(III) ion. Theoretical calculations can
help one to find the ligands that guarantee the most efficient energy transfer
to the Ln(III) ion and enable the design of highly efficient luminescence
materials. In this study, the influence of the ligand substituents on luminescent properties,
the molecular anisotropy and on the subsequent supramolecular organization of some Ln(III) complexes
was invesigated. Quantum-chemical methods were applied for the
simulation of equilibrium geometries, absorption, IR, NMR spectra and excited
states of some Ln(III) complexes with different ligand environment.
Theoretical
calculations of equilibrium geometries and IR spectra of Ln(III) complexes were performed in
the gas phase using the density functional theory and the exchange-correlation
functional PBE. NMR simulations were carried out by GIAO method. The energies
of the lowest singlet and triplet excited states were found by TDDFT method
(functionals PBE, B3LYP) in program Firefly 8. For Ln(III) ions the scalar relativistic 4f-in-core pseudopotentials with
the associated valence basis sets were used (ECP52MWB for Eu(III), ECP53MWB for
Gd(III), ECP54MWB for Tb(III) and ECP57MWB for Er(III)). For other atoms 6-31G(d,p)
basis set was applied. Absorption
spectra were calculated using SMLC model in ORCA 3.0.3 program.
It was established that the
coordination polyhedrons of the studied Ln(III) complexes were a square
antiprism and a dodecahedron. However, it should be mentioned that calculations
do not take into account the influence of neighboring molecules. Therefore the
geometric distortions that occur while molecules are packaging in crystal are
eliminated. Optimization of the Ln(III) complexes was accompanied by a slight
distortion of the coordination polyhedron due to the steric hindrance caused by
terminal substituents in the ligands. The isomer with a crosswise arrangement
of β-diketones in the complex when alkyl substitutes do not sterically
hinder each other was chosen for calculations because of its lowest energy. The
optimized structure of one of the studied Ln(III) complexes is shown in Fig. 1.
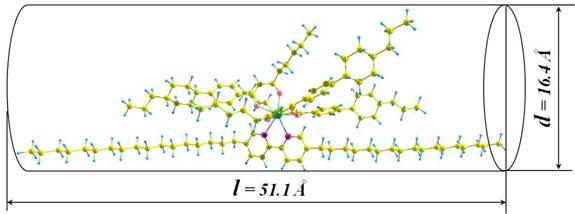
Fig. 1. Optimized structure and geometric parameters of
one of the studied Ln(III) complexes, where l
and d - length and width of the
molecule, respectively
The calculated UV, IR, NMR spectra
and the excited states were confirmed experimentally. Good agreement between simulated and experimental data showed that the
proposed methodology allows to predict the photophysical properties of Ln(III)
complexes and can be used to describe intramolecular energy transfer processes.
Correlations between the positions of the excited levels and the values
of absolute quantum yield were established, the main intramolecular energy
transfer channels were determined. Influence of the nature of substituents on luminescence
properties of the complexes and the effectiveness of their use in
optoelectronics was revealed. It was revealed that β-diketones play a
major role during the photoexcitation of the Ln(III) complexes since their
geometry considerably changes in comparison with other ligands. Quantum-chemical simulations discovered the
structural features of the Ln(III) compounds that predetermine their effective
luminescence and further application as luminescence functional materials.
The calculations were
performed using the facilities of the Joint Supercomputer Center of Russian
Academy of Sciences and the Supercomputing Center of Lomonosov, Moscow State
University [2]. This work was supported
by the grant of the President of the Russian Federation for the state support
of the young Russian scientists - candidates of sciences (No МК-7320.2016.3).
References:
1. Romanova
K.A., Freidzon A.Ya., Bagaturyants A.A., Galyametdinov Yu.G. Ab initio study of
energy transfer pathways in dinuclear lanthanide complex of europium(III) and
terbium(III) ions // Journal of Physical Chemistry A. 2014. V. 118. № 47. P.
11244-11252.
2.
Voevodin Vl.V., Zhumatiy S.A., Sobolev S.I., Antonov A.S., Bryzgalov P.A., Nikitenko D.A., Stefanov K.S., Voevodin Vad.V. Practice of “Lomonosov” Supercomputer // Open Systems
J. 2012. V. 7. P. 36-39.