Structure and function of
molecular chaperone HSP90
Jan Pałyga
and Łukasz Kozłowski
Department of Biochemistry and Genetics,
Świętokrzyska Academy, ul. Świętokrzyska 15,
25-406 Kielce, Poland
Introduction
Heat shock proteins (HSPs), an
important group of molecular chaperones, belong to a protein family whose
members can establish transitional complexes with virtually almost all proteins
produced in living cells. They assist in a polypeptide chain folding and
maintaining proper higher-order structures of client proteins. Using simple
models it has been suggested (Anfinsen,
1973; Dobson and Karplus, 1999) that a manner in which protein molecule can fold
depends on its amino acid sequence and that sequence itself is sufficient to
set up an active protein. This idea has been based on a premise that for a
given amino acid sequence there is only one highly preferred and energetically
most suitable state of the macromolecule (Fitzkee
et al., 2005; Vendruscolo et al., 2003). However, extrapolation of this view to other, often
extremely complex proteins, seemed to be hardly likely. In addition, a very
high concentration of proteins (up to 400g/L) inside the cell favouring
intermolecular interactions does not result in non-productive aggregation under
normal conditions (Ellis, 2001). Moreover, high protein concentrations at sites of
their production do not lead to non-desired interactions among nascent
polypeptide chains mediated by exposed hydrophobic residues and unstructered
segments. To counteract these sort of effects the cells have been equipped with
a webb of molecular chaperones assisting in proper polypeptide folding and
protein assembly. It has been shown (Dobson,
2003; Young et al., 2004) that the protein folding is strictly controlled from
the very beginning by a chaperone binding to nascent polypeptides to stabilize
their structure and/or prevent formation of premature protein assemblies with
inappropriate intra- and intermolecular interactions. In general, chaperones
represent a class of auxiliary proteins that could reversibly associate in a
stoichiometric manner with nascent or near native proteins to assist in their
correct folding wihout forming a part of the mature protein with which they
interact (Rutherford, 2003; Young et al., 2004).
Heat shock proteins,
discovered in the middle of twentieth century in ‘puffs’ of lampbrush
chromosomes from heat-shocked fruit flies Drosophila
melanogaster (Cossins, 1998), represent one of the most abundant cellular
proteins. The term heat shock proteins is somehow misleading and refers only to
one of the conditions under which these proteins could be induced. In fact, the
HSP synthesis is promoted by virtually all kind of stressors, including
oxidative stress and free radical damage, exposure to heavy metals, spontanous
mutations or chronic degenerative diseases. Thus, they should be more correctly
described as ‘stress proteins’ (Bagatell
and Whitesell, 2004; Cossins, 1998). The essential function of HSP proteins is to prevent
inappriopriate interactions within and between cellular proteins and/or to
restore a native structure in the proteins damaged by stress, and if these
measures fail, the HSPs may also facilitate degradation of misfolded proteins
by ubiquitin-proteasome pathway (McClellan
et al., 2005).
Roughly, three functional
groups of heat shock proteins could be distinguished: (i) a subgroup, including
members of Hsp70 family, which binds to nascent proteins on the ribosomes, (ii)
a group represented by HSP90 family members whose main function is to
facilitiate final folding stage of the near-mature proteins, and (iii) a group
of chaperones from Hsp104 family that can resolubilize aggregated polypeptides
and recycle them through the chaperone network (Young et al., 2004). A division of HSP proteins into six families
(HSP100, HSP90, HSP70, HSP60, HSP40 and small HSPs) is based on their molecular
masses in kilodaltons and a sequence homology within family members (Muchowski and Wacker, 2005). Most of the HSP proteins is already abundant in
normal unstressed cells but their level can increase in response to stressors.
This property as well as a high evolutionary conservation of the Hsp genes that are present in virtually
all prokaryotic and eukaryotic cells (Chen
et al., 2006) shows that they are engaged in crucial and
conservative cell functions.
The HSP90 chaperone family
constitutes up to 1-2% of the total cytosolic proteins and their abundance may
increase about twofold under stress conditions (Whitesell and Lindquist, 2005). Hsp90, a dimeric protein consisting of three domain
(N-terminal ATP-binding domain, a middle region and a C-terminal domain
involved in homodimerization) is featured by a capability of specific
interaction at a late stage of folding with a set of cellular proteins engaged
in regulatory or signalling pathways, such as transciption factors and protein
kinases (Buchner, 1999). HSP90 is
also involved in reactivation of inactivated or denatured proteins under
environmental stress conditions (Nathan
et al., 1997).
Structure
and diversity of HSP90 family
HSP90 has a strong tendency to
form dimers, mostly homodimers. Each HSP90 monomer has a complex structure
consisting of a negatively charged and variable in length middle region that is
flanked by conservative N- and C-terminal domains (Prodromou and Pearl, 2003). Hsp90 protomers have a parallel linear arrangenment
so that N-terminal domains are at one end of the dimer and the C-terminal
domains at the other (Fig. 1). The protomers make a left-handed helical twist
around the long axis of the dimer (Ali
et al., 2006). A 25-kDa N-terminal domain is a binding site for
both ATP and anticancer agents geldanamycin (GA) and radicicol. As revealed by
X-ray crystallography, the N-terminus co-crystalized with GA contained nine α-helices (four of them of 310 type) and an
antiparallel β-sheet of
eight strands folded together into an α+β sandwich arranged in two layers. A hydrophobic pocket, about 15 Å
deep and 10 Å in diameter, placed in the central part of N-terminal
domain is the site for ATP and GA binding (Prodromou
et al., 1997; Stebbins et al., 1997). ATP binding is followed by a conformational
alteration in the domain (McLaughlin
et al., 2004). Owing to an unconventional ATP-binding motif
sometimes referred to as a Bergerat fold, the HSP90 belongs to GHKL superfamily
(which includes DNA gyrase, HSP90, histidine kinases EnvZ and CheA and
DNA-mismatch-repair MutL protein) (Dutta
and Inouye, 2000). The N-domain is connected to a middle segment through
a loop with an antiparallel b-strands separated by a charged linker.
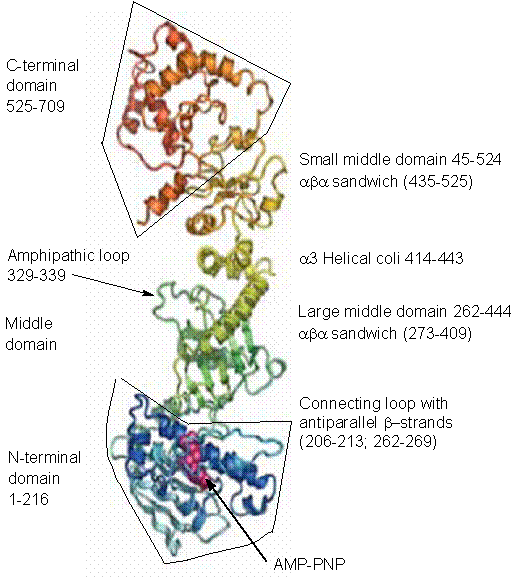
Fig. 1. An outline of monomer structure of yeast Hsp90 with details of
N-terminal, middle and C-terminal domains (Ali
et al., 2006; Pearl and Prodromou,
2006).
The middle domain with a large
three-layer αβα sandwich at its N-terminus connected to a smaller αβα domain at the C-terminus via an a3 helical coil is similar to that of dimeric GHKL
proteins (Meyer et al., 2003; Pearl and Prodromou, 2006). A ten-residue amphipathic loop interacting with
client proteins (Ali et al., 2006) projects from the inner face of the large αβα domain towards its counterpart in the other monomer.
An extended 25-residue loop links the middle segment with the beginning of a
curved a-helix at the start of the C-domain. A carboxy-terminal domain
tail-to-tail dimerization is important for the HSP90 function as only dimers
are fully active (Chadli et al., 2000).
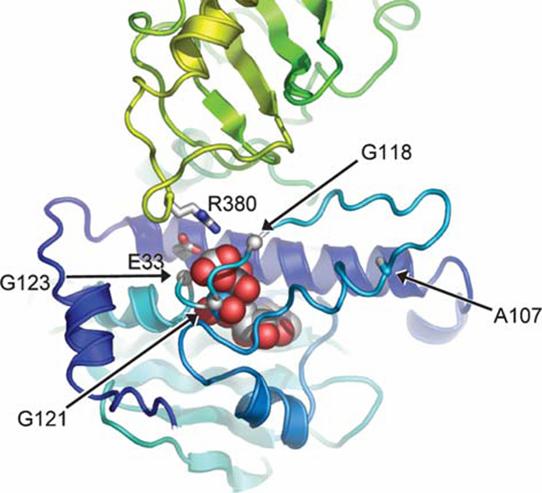
Fig. 2. Interaction around AMP-PNP in the full-length Hsp90 structure. A
lid segment bearing Ala107 fold over the nucleotide with the 118-GXXGXG-123
motif wrapping over the phosphates. Movement of the lid allows the Arg380 in
the catalytic loop from middle segment to come into contact with the g-phosphate of the AMP-PNP (Pearl
and Prodromou, 2006).
The dimer interface is formed
by a pair of helices from each monomer packed together to create a four-helix
bundle. At the very end of the C-terminus is located a conserved pentapeptide
MEEVD implicated in binding TPR-domain co-chaperons (D'Andrea and Regan, 2003). Although the main ATP-binding domain resides in the
N-terminus, there is evidence that the HSP90 C-terminus is harbouring a second
site of ATP binding (Marcu et al., 2000). The C-terminal domain could bind novobiocin and
cisplatin (Soti et al., 2002), and unexpectedly it possessed a weak but detectable
affinity for ADP and GTP (Soti et al., 2002) that could be discernible only after a prior ATP
binding to the N-terminal pocket. ATP-free HSP90 molecule assumes a bent
conformation in which C-terminal domain is in a close contact with a charged
region of the middle domain. Now, the only exposed motif available for
ATP-binding resides in the amino-terminal domain. ATP binding to this site
induces a conformational alteration which promotes binding of the lysine in the
middle domain with the ATP γ-phosphate group liberating C-terminal ATP-binding motif (Soti et al., 2002). Despite low ATP hydrolysis rate (Scheibel et al., 1998), the C-terminus seems to be indispensable for HSP90
function in vivo (Panaretou et al., 1998).
Detailed molecular
rearrangements during Hsp90 binding to its substrates and cochaperones have
been clarified by a crystal structure determination in a complex with
non-hydrolysable ATP analogue and the co-chaperone p23/Sba1 (Ali et
al., 2006). The N-terminal b-strand (residues 1-9) of each monomer crosses over to
make hydrogen bond to the edge of the main b-sheet in the N-terminal domain of the other monomer
with concomitant movement of the first a-helix (residues 13-22) so that the lid segment
(residues 94-125) swings nearly 180° to fold over the mouth of the nucleotide-binding
pocket. This event exposes a hydrophobic region that forms an interface with
which may interact a similar region of the adjacent monomer domain. Although
the middle domains move closer to each other, they still stay separated. Each
of the middle domains interact with the N-domain of the other monomer as well
as with its own N-domain. Now, the 118-GXXGXG123- motif at the C-terminal end
of the lid is wrapped around the b- and g-phosphates and distal parts of the lid interact with
ribose ring of the bound ATP (Fig. 2). In the middle segment, a catalytic loop
(residues 370-390) forming a short helix now unravels and extends down towards
the opening of the nucleotide-binding pocket in the N-terminal domain.
Activating cochaperone Aha1 promotes the mobility of this loop (Pearl and Prodromou, 2006).
Table
1. Members of HSP90 family.
|
Protein |
Species or cell compartment |
|
HSP90α |
Homo sapiens (major isoform) |
|
HSP90β |
Homo sapiens (minor isoform) |
|
HSP90N |
Homo sapiens |
|
HSP86 |
Mus musculus |
|
HSP84 |
Mus musculus |
|
HSP83 |
Drosphila melanogaster |
|
Hsc82 |
Saccharomyces cerevisiae |
|
HSP82 |
Saccharomyces cerevisiae |
|
HtpG |
Escherichia coli |
|
Grp94/gp96 |
Endoplasmic reticulum |
|
TRAP1(HSP75) |
Mitochondrial matrix |
|
cpHSP82 |
Chloroplasts |
HSP90 chaperons are members of
a large protein family (Table 1) present in bacteria, yeast and multicellular
organisms (Chen et al., 2006) with homologous proteins occurring not only in
cytoplasm but also in endoplasmic reticulum, mitochondria and chloroplasts (Buchner, 1999; Csermely et al., 1998). There are two main cytoplasmatic HSP90 isoforms: an
inducible HSP90α and
constitutively expressed HSP90β.
The isoform HSP90α is usually less abundant, but its amount can increase
under stress conditions. Slight differences in carboxy-terminal domain (Chen et al., 2005) are responsible for a weaker stability of ββ dimmers, as compared to αα dimers. At low abundance, both isoforms can also
exist as monomers, heterodimers αβ and oligomers. Although nucleotide sequences of both
forms are 76% identical and probably originated as a consequence of a gene
duplication about 500 milions years ago, their amino acid sequences are less
divergent, because differences are localised mostly in 5’ and 3’ untranslated
regions, promoters and introns (however, most eukaryotic Hsp genes do not contain introns) (Csermely
et al., 1998; Sreedhar et al., 2004). Recently, an aditional isoform HSP90N was detected (Grammatikakis et al., 2002). This isoform is a 75 kDa protein with a shortened
N-terminal domain consisting of 30 amino acids only and therefore deprived of
ATPase function.
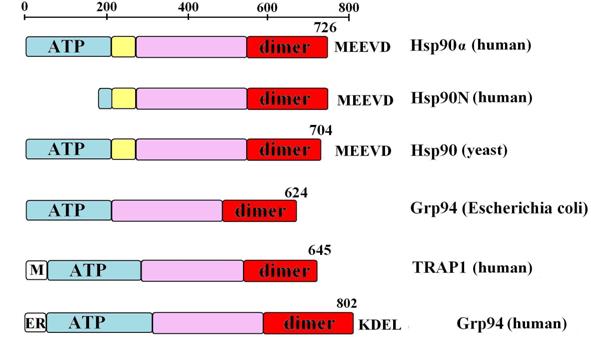
Figure
3. Domain structure of various HSP90 variants: ATP, ATP-binding N-terminal
domain (blue), charged region (yelow) with the rest of middle domain (purple)
and C-terminal dimerization domain (dimer, red). Mitochondrial and endoplasmic
reticulum signal sequences are indicated with ‘M’ and ‘ER’, respectively.
Conserved C-terminal amino acid sequences are shown in a single letter code.
The lenght of each protein is provided. Data are from (Buchner, 1999; Sreedhar et
al., 2004; Wegele et al., 2004).
The other members of HSP90
family are Grp94, TRAP1 (HSP75) and cpHSP82 (Table 1 and Figure 3) (Sreedhar et al., 2004; Young et al.,
2001). Chaperone Grp94 (glucose-regulated protein with
molecular mass of 94 kDa), the most abundant protein of the endoplasmic
reticulum (ER), is about 50% homologous to cytoplasmatic forms of HSP90,
whereas TRAP1 (TNF receptor-associated protein 1), localised in mitochondrial
matrix, is only 35% identical so that it exhibited less similarity to its
eukaryotic counterparts than to prokaryotic HSP90 homologue named HtpG). While
the function of cytoplasmatic forms of HSP90 is quite broad, the organellar
members of the family, restricted to higher eukaryotes, are more specialized (Picard, 2002).
Table 2. A partial list of cochaperones and
representatives of HSP90 client proteins (a full list is available at http://www.picard.ch/)
|
Cochaperones: Aha1 and its homologue Hch1; Cdc37/p50 and its homologue Harc Hsj, human DnaJ homologue HSP70 (Ssa1p); HSP40 (Hdj1, Ydj1p); Sba1/p23 Proteins with TPR motifs: CHIP (an
E3/E4-ubiquitin ligase required for proteasome-targeted destruction); Cns1(cyclophilin seven supressor 1); Cyclophilin
Cyp40 (Cpr6, Cpr7) and immunophilins FKBP51, FKBP52 ( peptidyl-prolyl
isomerases); HOP (p60, Sti1); PP5 phosphatase (Ser/Thr
protein phosphatase). |
|
Client proteins: Transcription
factors: Steroid hormone receptors: androgen, estrogen, glucocorticoid,
mineralcorticoid and progesteron receptors; HSF-1; IRF2; p53; PPARa and b; STAT3; Kinases: Akt/PTB; Aurora B;
Bcr-Abl; casein kinase II; Cdk2; Cdk4; Cdk6; Cdc9; Chk1 (checkpoint kinase
1); ErbB2; Insulin receptor; JAK1; MEK; c-Mos; PKCl; Polo;
Raf-1, RET/PTC1; SRC-related; Wee1; Other: calcineurin;
calmodulin; CFTR; cytoskeletal proteins: actin, myosin, tubulin; DNA
polimerase a; G
protein (subunits α0, α12, βγ); HDAC6; Histones
H1, H2A, H2B, H3 and H4; MRE11/Rad50/NBS1; MMP2 (matrix metalloproteinase 2); MTG8;
Neuropeptide Y; Prolactin receptor; Proteasome; Smyd3; SV40 large T-antigen; a-Synuclein;
Tau protein; Telomerase; Vimentin. |
HSP90
substrates
Heat shock protein 90 is able
to interact with an enormous number of highly specialized cellular substrate
proteins called client proteins (Table 2) among which are steroid hormone
receptors (SHR), transcription factors and plethora of kinases and other
cellular proteins (Wegele et al., 2004). A formation of compact form of HSP90 dimer is
directly controlled by ATP/ADP deposition and co-chaperone binding and most of
interactions between HSP90 and substrates are localized on external interfaces
formed during conformational arrangements (Ali
et al., 2006; Shiau et al., 2006). Although HSP90 do not exhibit any specificity to a
particular amino acid motif, it can recognize
its client proteins in a specific manner.
HSP90 is an indispensable
agent for activation of viral kinase p60v-src that in its active
state is attached to the plasma membrane and causes oncogenic proliferation of
infected cells. Initially, kinase p60v-src is a soluble protein with
a high affinity to HSP90. In this complex it is subjected to phosphorylation
and myristylation and is finally transported in a direct proximity of plasma
membrane where it is released (Buchner,
1999; Yahara, 1999). By contrast, its close homolog p60 c-src,
a constituent of regular cells, is far less dependent on HSP90 (Sangster et al.,
2004).
Most of the HSP90 clients are
transcription factors, housekeeping proteins or protein components of various
signaling pathways controlling development and basic function of the living
cells (Table 2). Little is known about a specificity of the HSP90 isoforms. The
chaperone HSP90N can specificially interact with a Raf kinase and translocate
it to a plasma membrane independently of c-Ras pathway (Grammatikakis et al.,
2002). Although cytosolic HSP90 isoforms could also
activate Raf kinases (van der
Straten et al., 1997) it is believed (Grammatikakis
et al., 2002) that most of their activity is owned to HSP90N.
Taken together, HSP90s are
unique among other heat shock proteins most of which, including the best
examined HSP70, associate with their clients already during their synthesis or
when they recognize hydrophobic residues on the surface of damaged proteins. In
contrast to other HSPs, the clients of HSP90 are highly specific near-native
metastable proteins (Wegele et al., 2004) . Usually, the interaction of HSP90 with its client
poteins is involved in their translocation, activation or/and stabilization (Young et al., 2003). Unfortunately, we do not know what determines the
affinity of HSP90 to each protein and as noted above no specific sequence
motifs have been identified so far.
HSP90
co-chaperones
In addition to its clients, HSP90
interacts with a special group of proteins including other chaperones which can
form multiprotein complexes. These proteins are referred to as co-chaperons
(Table 2). A make-up of these complexes is diverse and directly determines the
kind of interacting clients. Most of the co-chaperones harbour TPR
(tetratricopeptide repeat) domains, 34-amino-acid degenerate repeat sequences,
with help of which the proteins interacts with each other (D'Andrea and Regan, 2003; Young et al., 2004). TPR domains have a high affinity to carboxy-terminal
pentapeptide MEEVD of the HSP90 (Fig. 3). A similar motif EEVD can be found at
HSP70 C-terminus (Wegele et al., 2004). This highly conserved sequence contributes in a significant
way to the affinity of HSP90 to its TPR-containing co-chaperones (Pearl and Prodromou, 2006).
The best studied example of
the cooperation between HSP90 and other co-chaperones is activation of steroid
hormone receptors (SHRs) (Grad and
Picard, 2007; Pratt and Toft, 2003). In the absence of ligand the steroid hormone
receptors are bound into complexes containing HSP90. The HSP90 chaperone allows the receptor to achieve a
structural conformation that is competent for ligand binding, nuclear
translocation and consequently, gene regulation (Kovacs et al.,
2005a). The
chaperone multiprotein complex is changing in a cyclic manner so that the
successive auxiliary proteins (mainly co-chaperones) associate and dissociate
in due time from the complex after fullfilling their function. In addition, the
whole process is depended on and regulated by ATP (Fig. 4). The minimal complex
indispensable for SHR activation encompasses HSP40, HSP70, HOP (Hsp organizing protein),
HSP90 and p23/Sba1 protein (Kosano
et al., 1998). Initially, just after completion its translation,
the steroid hormone receptor binds to HSP40 and HSP70 (Fig. 4a). Then it is
donated to the HSP90 through interaction with HOP protein (Wegele et al., 2004) that is almost entirely composed of TPR domains and
serves as a linker between HSP70 and HSP90 connecting them by their
carboxy-terminal domains (Odunuga et al., 2004). This transfer takes place only if ADP is bound to
HSP90. The exchange of ADP to ATP inside N-terminal pocket induces dissociation
of HSP70 and its co-chaperones from the complex that now is free to associate
with protein p23/Sba1, which binds to the N-terminal side of HSP90 dimer and
prevents ATP hydrolysis (Ali et al., 2006; McLaughlin et al., 2002), and immunophilin, which generally substitutes for
HOP (Fig. 4). Until recently, the function of immunophilins FKBP51 (FK-506-binding protein 51) and FKBP52 with peptidyl-prolyl cis–trans isomerase activity (Davies and Sanchez, 2005) that can catalyze
the conversion of prolyl–peptide bonds from trans- to cis-proline,
often a rate-limiting step in protein folding, was little known. The name for FKBPs derives from their ability to bind
immunosuppressive drug FK-506 (Davies
and Sanchez, 2005). Recently, it has been reported (Pratt et al., 2004) that they are responsible for transportation of
HSP90-SHR-ligand complexes along the microtubule fibers. In this way a
translocation of hormones (Davies
and Sanchez, 2005; Pratt et al.,
2004), p53 protein (Galigniana
et al., 2004) and probably other HSP90 substrate proteins within
cytoplasm is fast and tightly controlled. ATP hydrolysis inside HSP90
nucleotide-binding pocket leads to the dissociation of the complex, and
liganded steroid hormone receptors dimerize and are translocated to the nucleus
(Fig. 4d). Subsequently, SHR-hormone complexes bind to particular DNA sequences
in the promoters of hormone-responsive genes to control their transcription.
Remarkably, the movement of SHRs inside the nucleus is also HSP90- and
ATP-dependent (Elbi et al., 2004). Details of the translocation of liganded SHRs are
still unclear (Grad and Picard,
2007) and likely depend on HSP90-HSP70 complexes that could
be transmitted through the nuclear envelope pores as a whole to deliver the
signal to the nuclear interior in a direct vicinity of the chromatin (Pratt et al., 2004). But on the other hand an opposite scenario in which
steroid hormone receptors could shuttle between separate HSP90 molecular
complexes on both sides of the nuclear envelope is also possible (Elbi et al., 2004).
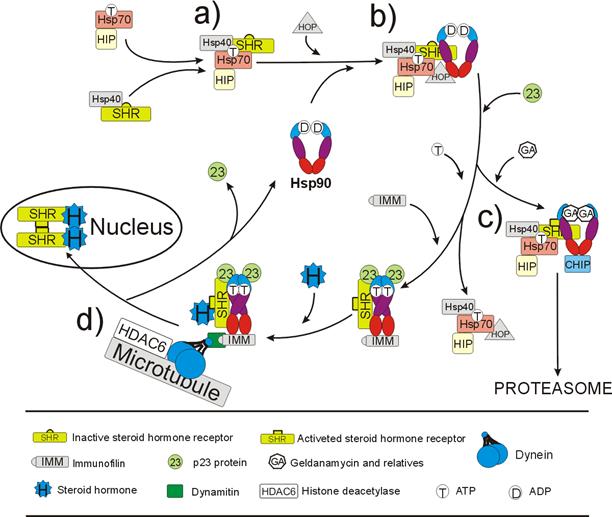
Fig. 4. HSP90-dependent cycle
of steroid hormone receptor (SHR) activation. a) Establishing HSP70-SHR
complex; b) transmission of SHR onto HSP90 dimer with the help of HSP70 and
HOP; c) association of p23/Sba1 rearranges HSP90 conformation. Now, if the
chaperone binds geldanamycin, which mimics ADP binding, proteins p23 and HOP
dissociate. CHIP, an E3 ubiqutin ligase, is attached to the complex and SHR
receptor is being degraded through the proteasome-mediateted pathway. On the
other hand, if HSP90 binds ATP, the HSP70 and its co-chaperones dissociate
giving the place for immunophilins (FKBP51, which is substituted following
ligand binding by FKBP52); d) HSP90-immunophilin-p23 complex activates SHR,
which can now bind a steroid hormone. After that the complex binds to dynamitin
and dynein, the microtubule-associated proteins, and is moving along
cytoskeleton structures towards nucleus. Liganded SHRs dimerize and interact
with promoter sequences of target genes. For simplicity, the HDAC6 influence
has been ommited. See text for details.
As mentioned above, the ATPase
function of HSP90 is crucial for determining its full activity. If ADP/ATP
binding pocket is occupied by inhibitory agents mimicking nucleotide structure
(radicicol, geldanamycin and their relatives), the client protein cannot
dissociate from the chaperone complex and will bind CHIP, an E3 ubiquitin
ligase, which stimulates proteasomal degradation of the client protein (Fig.
4c) (Chiosis et al., 2004; Cyr et al.,
2002; Neckers and Neckers, 2005).
A large set of HSP clients are
protein kinases (Table 2). The interaction between Hsp90 and kinases is
facilitated by Cdc37 chaperone (Caplan
et al., 2007). The Cdc37 binds both to a catalytic domain of the
kinase and to the N-terminal domain of HSP90. Cdc37 dimers localized between
N-termini and the charged region of middle domains of HSP90 prevent ATP
hydrolysis, as p23/Sba1 does (Roe et al., 2004; Zhang et al., 2004). Additionally, HSP90-Cdc37-kinase complexes include
other specific proteins like HOP, Aha1 and protein phosphatase PP5 (Pearl, 2005).
Both HSP90 structure and
ATPase activity are influenced by co-chaperone binding. Initially, ATP
hydrolysis rate is almost undetectable but increases up to 200-fold after
binding of glucocorticoid receptor (Pratt
et al., 2004). The co-chaperons HOP, p23/Sba1, Aha1 and others act
as HSP90 supressors or activators that regulate a shift in its structure or/and
ATPase activity (Pearl and
Prodromou, 2006).
Recent investigations indicate
that a reversible acetylation of HSP90 is a key factor in controling its
function. Yu and co-workers (Yu et al., 2002) established that HSP90 is one of many substrates for
histone deacetylase HDAC6. Inactivation of HDACs using specific inhibitors (Bali et al., 2005), RNA interference (Murphy
et al., 2005) or mutational analysis (Scroggins et al.,
2007) leads to hyperacetylation of HSP90, including a
conserved K294 in the middle domain (Scroggins
et al., 2007), and rapid disassembly of the multiprotein complexes (Murphy et al., 2005) or even proteosomal degradation of clients such as
Raf-1, AKT, Bcl-Abl and p53 proteins (Bali
et al., 2005; Yu et al., 2002). Hyperacetylated HSP90 forms only short-lived
complexes incapable of effective and stable SHR ligand binding, nuclear
translocation and gene activation (Murphy
et al., 2005); (Kovacs
et al., 2005b). Acetylated HSP90-SHR complex has a low affinity to
ATP and p23/Sba1 co-chaperone what prevents futher remodeling and SHR receptor
activation (Murphy et al., 2005). HDAC6 is a unique histone deacetylase because of its
cytoplasmatic localisation where it associates with microtubules to deacetylate
α-tubulin (Boyault et al., 2007). Presumably, HDAC6 keeps a low level of HSP90
acetylation during the translocation of the complex along microtubules (Fig.
4d). Although an enzyme acetylating HSP90 protein has not been revealed yet (Scroggins et al., 2007), it shoud be localised in the vicinity or inside the
nucleus. HSP90 appears to
become transiently acetylated upon receptor activation after ligand stimulation
(Kovacs et al., 2005a). HSP90 acetylation might allow the conversion of
SHR-HSP90 from a stable complex into a dynamic one by dissociating p23/Sba1 from
the HSP90 complex, thereby enabling SHR to enter the nucleus for
transcriptional activation. As acetylated HSP90 exhibits reduced binding toward
SHR and p23/Sba1, the subsequent deacetylation by HDAC6 would then allow HSP90
to restore the productive chaperone complex (Kovacs
et al., 2005a). Therefore,
HSP90 acetylation may represent a regulatory signal triggering a discharge of
the SHR-HSP90 complexes. After SHR release, the deacetylase HDAC6 may sneak
again to the complex to restore a low level of HSP90 acetylation.
Although HSP90 function seems
to be inseparably associated with a large number of chaperones and other
proteins, it turned out that HSP90 can go without co-chaperones and retain its
chaperone activity outside the cell as well. In some cases HSP90 is expressed
extracellularly where it interacts with MMP2 (matrix metalloproteinase 2), an
enzyme involved in spreading and invasion of tumour cell (Eustace et al., 2004). Most interestingly, only HSP90α isoform, but not HSP90β, take part in this process. This is a direct evidence
for functional diversification among HSP90 variants (Eustace et al.,
2004).
HSP90
function
A basic HSP90 function, shared by all
heat shock proteins, is a protection (‘chaperoning’) against a loss of activity
by other proteins under stress conditions. Under unfavourable circumstances,
the proteins tend to unfold and/or aggregate . Heat shock proteins including
HSP90 target conformationally altered polypeptides to restore their proper
native structure. A cursory glance at the list of HSP90 client proteins (Table
2) shows that a majority of substrate proteins is engaged in multiple signaling
pathways (Rutherford et al., 2007b; Soti et al., 2005; Zhao and Houry, 2007), chromatin transactions and transcriptional
regulation (Wong and Houry, 2006;
Zhao and Houry, 2005), cell cycle regulation (Burrows et al.,
2004) and malignant growth (Beliakoff and Whitesell, 2004; Neckers, 2007). These bizarre activities could reflect a need for
both quick and accurate response to external and internal stimuli. This can be
achieved only through extremely precise transduction of individual information
signals through a cellular signalling network needed for cell cycle progresion
and regulation, and successful completion of development. The interactions of
kinases, nuclear receptors and transcription factors with HSP90 enable
achieving a proper and precise folding and maturation as well as movement of
signal molecules to their destination within the cell (Richter and Buchner, 2001). Additionally, in last years the heat shock proteins,
including HSP90, were implicated in immune responses as well (Gullo and Teoh, 2004; Nardai et al., 2006).
Although HSP90 chaperone is
only one of many HSP proteins, it is an indispensable cellular protein given
its ubiquitous occurrence, high evolutionary conservation and a type and number
of HSP90 client proteins. In fact, detrimental effects of the lack of HSP90 was
experimentally confirmed in yeast S.
cerevisiae (Borkovich et al., 1989), nematode Caenorhabditis
elegants (Birnby et al., 2000) and fruifly D.
melanogaster (van der Straten et al., 1997) deprived of Hsp90
genes. In mice, homozygous idviduals with only one mutated gene coding
HSP90β isoform, in the presence of normal HSP90a counterpart, did not develop placental labyrinth and
died at early embryonic stages (Voss
et al., 2000).
HSP90 through interactions
with specific set of proteins can act as a genetic capacitor during exposure to
stressful conditions in Drosophila (Carey
et al., 2006; Debat et al., 2006; Milton et al., 2006; Rutherford and Lindquist,
1998), Arabidopsis
thaliana (Queitsch et al., 2002; Sangster et al., 2007; Sangster et al., 2004; Sangster and Queitsch,
2005) and fish Danio rerio (Yeyati et al.,
2007). There is a growing body of fossil records
suggesting that organisms could have evolved in a rapid and step-wise manner,
as opposite to a well established Darwinian model of gradual changes (Cossins, 1998). Genetic capacitors moderate expression of heritable
variation and provide a mechanism for rapid evolution (Rutherford et al.,
2007a; Rutherford, 2003) through stress-sensitive storage and release of
genetic variation to facilitate adaptive evolution in unpredictabe
environments.
References
Ali M. M., Roe S. M., Vaughan C. K.,
Meyer P., Panaretou B., Piper P. W., Prodromou C. and Pearl L. H. (2006).
Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 440, 1013-1017.
Anfinsen C. B. (1973). Principles
that govern the folding of protein chains. Science
181, 223-230.
Bagatell R. and Whitesell L. (2004).
Altered Hsp90 function in cancer: a unique therapeutic opportunity. Mol Cancer Ther 3, 1021-1030.
Bali P., Pranpat M., Bradner J.,
Balasis M., Fiskus W., Guo F., Rocha K., Kumaraswamy S., Boyapalle S., Atadja
P., Seto E. and Bhalla K. (2005). Inhibition of histone deacetylase 6
acetylates and disrupts the chaperone function of heat shock protein 90: a
novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem 280, 26729-26734.
Beliakoff J. and Whitesell L.
(2004). Hsp90: an emerging target for breast cancer therapy. Anticancer Drugs 15, 651-662.
Birnby D. A., Link E. M., Vowels J.
J., Tian H., Colacurcio P. L. and Thomas J. H. (2000). A transmembrane guanylyl
cyclase (DAF-11) and Hsp90 (DAF-21) regulate a common set of chemosensory
behaviors in caenorhabditis elegans. Genetics
155, 85-104.
Borkovich K. A., Farrelly F. W.,
Finkelstein D. B., Taulien J. and Lindquist S. (1989). hsp82 is an essential
protein that is required in higher concentrations for growth of cells at higher
temperatures. Mol Cell Biol 9, 3919-3930.
Boyault C., Sadoul K., Pabion M. and
Khochbin S. (2007). HDAC6, at the crossroads between cytoskeleton and cell
signaling by acetylation and ubiquitination. Oncogene 26, 5468-5476.
Buchner J. (1999). Hsp90 & Co. -
a holding for folding. Trends Biochem Sci
24, 136-141.
Burrows F., Zhang H. and Kamal A.
(2004). Hsp90 activation and cell cycle regulation. Cell Cycle 3, 1530-1536.
Caplan A. J., Mandal A. K. and
Theodoraki M. A. (2007). Molecular chaperones and protein kinase quality
control. Trends Cell Biol 17, 87-92.
Carey C. C., Gorman K. F. and
Rutherford S. (2006). Modularity and intrinsic evolvability of hsp90-buffered
change. PLoS ONE 1, e76.
Chadli A., Bouhouche I., Sullivan
W., Stensgard B., McMahon N., Catelli M. G. and Toft D. O. (2000). Dimerization
and N-terminal domain proximity underlie the function of the molecular
chaperone heat shock protein 90. Proc
Natl Acad Sci U S A 97,
12524-12529.
Chen B., Piel W. H., Gui L., Bruford
E. and Monteiro A. (2005). The HSP90 family of genes in the human genome:
Insights into their divergence and evolution. Genomics 86, 627-637.
Chen B., Zhong D. and Monteiro A.
(2006). Comparative genomics and evolution of the HSP90 family of genes across
all kingdoms of organisms. BMC Genomics
7, 156.
Chiosis G., Vilenchik M., Kim J. and
Solit D. (2004). Hsp90: the vulnerable chaperone. Drug Discov Today 9,
881-888.
Cossins A. (1998). Cryptic clues
revealed. Nature 396, 309-310.
Csermely P., Schnaider T., Soti C.,
Prohaszka Z. and Nardai G. (1998). The 90-kDa molecular chaperone family:
structure, function, and clinical applications. A comprehensive review. Pharmacol Ther 79, 129-168.
Cyr D. M., Hohfeld J. and Patterson
C. (2002). Protein quality control: U-box-containing E3 ubiquitin ligases join
the fold. Trends Biochem Sci 27, 368-375.
D'Andrea L. D. and Regan L. (2003).
TPR proteins: the versatile helix. Trends
Biochem Sci 28, 655-662.
Davies T. H. and Sanchez E. R.
(2005). FKBP52. Int J Biochem Cell Biol
37, 42-47.
Debat V., Milton C. C., Rutherford
S., Klingenberg C. P. and Hoffmann A. A. (2006). Hsp90 and the quantitative
variation of wing shape in Drosophila
melanogaster. Evolution Int J Org
Evolution 60, 2529-2538.
Dobson C. M. (2003). Protein folding
and misfolding. Nature 426, 884-890.
Dobson C. M. and Karplus M. (1999).
The fundamentals of protein folding: bringing together theory and experiment. Curr Opin Struct Biol 9, 92-101.
Dutta R. and Inouye M. (2000). GHKL,
an emergent ATPase/kinase superfamily. Trends
Biochem Sci 25, 24-28.
Elbi C., Walker D. A., Romero G.,
Sullivan W. P., Toft D. O., Hager G. L. and DeFranco D. B. (2004). Molecular
chaperones function as steroid receptor nuclear mobility factors. Proc Natl Acad Sci U S A 101, 2876-2881.
Ellis R. J. (2001). Macromolecular
crowding: obvious but underappreciated. Trends
Biochem Sci 26, 597-604.
Eustace B. K., Sakurai T., Stewart
J. K., Yimlamai D., Unger C., Zehetmeier C., Lain B., Torella C., Henning S.
W., Beste G., Scroggins B. T., Neckers L., Ilag L. L. and Jay D. G. (2004).
Functional proteomic screens reveal an essential extracellular role for hsp90
alpha in cancer cell invasiveness. Nat
Cell Biol 6, 507-514.
Fitzkee N. C., Fleming P. J., Gong
H., Panasik N., Jr., Street T. O. and Rose G. D. (2005). Are proteins made from
a limited parts list? Trends Biochem Sci
30, 73-80.
Galigniana M. D., Harrell J. M.,
O'Hagen H. M., Ljungman M. and Pratt W. B. (2004). Hsp90-binding immunophilins
link p53 to dynein during p53 transport to the nucleus. J Biol Chem 279,
22483-22489.
Grad I. and Picard D. (2007). The
glucocorticoid responses are shaped by molecular chaperones. Mol Cell Endocrinol 2, 2.
Grammatikakis N., Vultur A., Ramana
C. V., Siganou A., Schweinfest C. W., Watson D. K. and Raptis L. (2002). The
role of Hsp90N, a new member of the Hsp90 family, in signal transduction and
neoplastic transformation. J Biol Chem
277, 8312-8320.
Gullo C. A. and Teoh G. (2004). Heat
shock proteins: to present or not, that is the question. Immunol Lett 94, 1-10.
Kosano H., Stensgard B.,
Charlesworth M. C., McMahon N. and Toft D. (1998). The assembly of progesterone
receptor-hsp90 complexes using purified proteins. J Biol Chem 273,
32973-32979.
Kovacs J. J., Cohen T. J. and Yao T.
P. (2005a). Chaperoning steroid hormone signaling via reversible acetylation. Nucl Recept Signal 3, e004.
Kovacs J. J., Murphy P. J., Gaillard
S., Zhao X., Wu J. T., Nicchitta C. V., Yoshida M., Toft D. O., Pratt W. B. and
Yao T. P. (2005b). HDAC6 regulates Hsp90 acetylation and chaperone-dependent
activation of glucocorticoid receptor. Mol
Cell 18, 601-607.
Marcu M. G., Chadli A., Bouhouche
I., Catelli M. and Neckers L. M. (2000). The heat shock protein 90 antagonist
novobiocin interacts with a previously unrecognized ATP-binding domain in the
carboxyl terminus of the chaperone. J
Biol Chem 275, 37181-37186.
McClellan A. J., Tam S., Kaganovich
D. and Frydman J. (2005). Protein quality control: chaperones culling corrupt
conformations. Nat Cell Biol 7, 736-741.
McLaughlin S. H., Smith H. W. and
Jackson S. E. (2002). Stimulation of the weak ATPase activity of human hsp90 by
a client protein. J Mol Biol 315, 787-798.
McLaughlin S. H., Ventouras L. A.,
Lobbezoo B. and Jackson S. E. (2004). Independent ATPase activity of Hsp90
subunits creates a flexible assembly platform. J Mol Biol 344, 813-826.
Meyer P., Prodromou C., Hu B.,
Vaughan C., Roe S. M., Panaretou B., Piper P. W. and Pearl L. H. (2003).
Structural and functional analysis of the middle segment of hsp90: implications
for ATP hydrolysis and client protein and cochaperone interactions. Mol Cell 11, 647-658.
Milton C. C., Ulane C. M. and
Rutherford S. (2006). Control of canalization and evolvability by hsp90. PLoS ONE 1, e75.
Muchowski P. J. and Wacker J. L.
(2005). Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 6, 11-22.
Murphy P. J., Morishima Y., Kovacs
J. J., Yao T. P. and Pratt W. B. (2005). Regulation of the dynamics of hsp90
action on the glucocorticoid receptor by acetylation/deacetylation of the
chaperone. J Biol Chem 280, 33792-33799.
Nardai G., Vegh E. M., Prohaszka Z.
and Csermely P. (2006). Chaperone-related immune dysfunction: an emergent
property of distorted chaperone networks. Trends
Immunol 27, 74-79.
Nathan D. F., Vos M. H. and
Lindquist S. (1997). In vivo functions of the Saccharomyces cerevisiae Hsp90
chaperone. Proc Natl Acad Sci U S A 94, 12949-12956.
Neckers L. (2007). Heat shock
protein 90: the cancer chaperone. J Biosci
32, 517-530.
Neckers L. and Neckers K. (2005).
Heat-shock protein 90 inhibitors as novel cancer chemotherapeutics - an update.
Expert Opin Emerg Drugs 10, 137-149.
Odunuga O. O., Longshaw V. M. and
Blatch G. L. (2004). Hop: more than an Hsp70/Hsp90 adaptor protein. Bioessays 26, 1058.
Panaretou B., Prodromou C., Roe S.
M., O'Brien R., Ladbury J. E., Piper P. W. and Pearl L. H. (1998). ATP binding
and hydrolysis are essential to the function of the Hsp90 molecular chaperone
in vivo. Embo J 17, 4829-4836.
Pearl L. H. (2005). Hsp90 and Cdc37
- a chaperone cancer conspiracy. Curr
Opin Genet Dev 15, 55-61.
Pearl L. H. and Prodromou C. (2006).
Structure and mechanism of the hsp90 molecular chaperone machinery. Annu Rev Biochem 75, 271-294.
Picard D. (2002). Heat-shock protein
90, a chaperone for folding and regulation. Cell
Mol Life Sci 59, 1640-1648.
Pratt W. B., Galigniana M. D.,
Harrell J. M. and DeFranco D. B. (2004). Role of hsp90 and the hsp90-binding
immunophilins in signalling protein movement. Cell Signal 16, 857-872.
Pratt W. B. and Toft D. O. (2003).
Regulation of signaling protein function and trafficking by the
hsp90/hsp70-based chaperone machinery. Exp
Biol Med (Maywood) 228, 111-133.
Prodromou C. and Pearl L. H. (2003).
Structure and functional relationships of Hsp90. Curr Cancer Drug Targets 3,
301-323.
Prodromou C., Roe S. M., O'Brien R.,
Ladbury J. E., Piper P. W. and Pearl L. H. (1997). Identification and
structural characterization of the ATP/ADP-binding site in the Hsp90 molecular
chaperone. Cell 90, 65-75.
Queitsch C., Sangster T. A. and
Lindquist S. (2002). Hsp90 as a capacitor of phenotypic variation. Nature 417, 618-624.
Richter K. and Buchner J. (2001).
Hsp90: chaperoning signal transduction. J
Cell Physiol 188, 281-290.
Roe S. M., Ali M. M., Meyer P.,
Vaughan C. K., Panaretou B., Piper P. W., Prodromou C. and Pearl L. H. (2004).
The Mechanism of Hsp90 regulation by the protein kinase-specific cochaperone
p50(cdc37). Cell 116, 87-98.
Rutherford S., Hirate Y. and Swalla
B. J. (2007a). The hsp90 capacitor, developmental remodeling, and evolution:
the robustness of gene networks and the curious evolvability of metamorphosis. Crit Rev Biochem Mol Biol 42, 355-372.
Rutherford S., Knapp J. R. and
Csermely P. (2007b). Hsp90 and developmental networks. Adv Exp Med Biol 594,
190-197.
Rutherford S. L. (2003). Between
genotype and phenotype: protein chaperones and evolvability. Nat Rev Genet 4, 263-274.
Rutherford S. L. and Lindquist S.
(1998). Hsp90 as a capacitor for morphological evolution. Nature 396, 336-342.
Sangster T. A., Bahrami A., Wilczek
A., Watanabe E., Schellenberg K., McLellan C., Kelley A., Kong S. W., Queitsch
C. and Lindquist S. (2007). Phenotypic diversity and altered environmental
plasticity in Arabidopsis thaliana with reduced Hsp90 levels. PLoS ONE 2, e648.
Sangster T. A., Lindquist S. and
Queitsch C. (2004). Under cover: causes, effects and implications of
Hsp90-mediated genetic capacitance. Bioessays
26, 348-362.
Sangster T. A. and Queitsch C.
(2005). The HSP90 chaperone complex, an emerging force in plant development and
phenotypic plasticity. Curr Opin Plant
Biol 8, 86-92.
Scheibel T., Weikl T. and Buchner J.
(1998). Two chaperone sites in Hsp90 differing in substrate specificity and ATP
dependence. Proc Natl Acad Sci U S A 95, 1495-1499.
Scroggins B. T., Robzyk K., Wang D.,
Marcu M. G., Tsutsumi S., Beebe K., Cotter R. J., Felts S., Toft D., Karnitz
L., Rosen N. and Neckers L. (2007). An acetylation site in the middle domain of
hsp90 regulates chaperone function. Mol
Cell 25, 151-159.
Shiau A. K., Harris S. F.,
Southworth D. R. and Agard D. A. (2006). Structural Analysis of E. coli hsp90
reveals dramatic nucleotide-dependent conformational rearrangements. Cell 127, 329-340.
Soti C., Pal C., Papp B. and
Csermely P. (2005). Molecular chaperones as regulatory elements of cellular
networks. Curr Opin Cell Biol 17, 210-215.
Soti C., Racz A. and Csermely P.
(2002). A Nucleotide-dependent molecular switch controls ATP binding at the
C-terminal domain of Hsp90. N-terminal nucleotide binding unmasks a C-terminal
binding pocket. J Biol Chem 277, 7066-7075.
Sreedhar A. S., Kalmar E., Csermely
P. and Shen Y. F. (2004). Hsp90 isoforms: functions, expression and clinical
importance. FEBS Lett 562, 11-15.
Stebbins C. E., Russo A. A.,
Schneider C., Rosen N., Hartl F. U. and Pavletich N. P. (1997). Crystal
structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by
an antitumor agent. Cell 89, 239-250.
van der Straten A., Rommel C.,
Dickson B. and Hafen E. (1997). The heat shock protein 83 (Hsp83) is required
for Raf-mediated signalling in Drosophila. Embo
J 16, 1961-1969.
Vendruscolo M., Zurdo J., MacPhee C.
E. and Dobson C. M. (2003). Protein folding and misfolding: a paradigm of
self-assembly and regulation in complex biological systems. Philos Transact A Math Phys Eng Sci 361, 1205-1222.
Voss A. K., Thomas T. and Gruss P.
(2000). Mice lacking HSP90beta fail to develop a placental labyrinth. Development 127, 1-11.
Wegele H., Muller L. and Buchner J.
(2004). Hsp70 and Hsp90--a relay team for protein folding. Rev Physiol Biochem Pharmacol 151,
1-44. Epub 2004 Jan 2023.
Whitesell L. and Lindquist S. L.
(2005). HSP90 and the chaperoning of cancer. Nat Rev Cancer 5,
761-772.
Wong K. S. and Houry W. A. (2006).
Hsp90 at the crossroads of genetics and epigenetics. Cell Res 16, 742-749.
Yahara I. (1999). The role of HSP90
in evolution. Genes Cells 4, 375-379.
Yeyati P. L., Bancewicz R. M., Maule
J. and van Heyningen V. (2007). Hsp90 selectively modulates phenotype in
vertebrate development. PLoS Genet 3, e43.
Young J. C., Agashe V. R., Siegers
K. and Hartl F. U. (2004). Pathways of chaperone-mediated protein folding in
the cytosol. Nat Rev Mol Cell Biol 5, 781-791.
Young J. C., Barral J. M. and Ulrich
Hartl F. (2003). More than folding: localized functions of cytosolic
chaperones. Trends Biochem Sci 28, 541-547.
Young J. C., Moarefi I. and Hartl F.
U. (2001). Hsp90: a specialized but essential protein-folding tool. J Cell Biol 154, 267-273.
Yu X., Guo Z. S., Marcu M. G.,
Neckers L., Nguyen D. M., Chen G. A. and Schrump D. S. (2002). Modulation of
p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide
FR901228. J Natl Cancer Inst 94, 504-513.
Zhang W., Hirshberg M., McLaughlin
S. H., Lazar G. A., Grossmann J. G., Nielsen P. R., Sobott F., Robinson C. V.,
Jackson S. E. and Laue E. D. (2004). Biochemical and structural studies of the
interaction of Cdc37 with Hsp90. J Mol
Biol 340, 891-907.
Zhao R. and Houry W. A. (2005).
Hsp90: a chaperone for protein folding and gene regulation. Biochem Cell Biol 83, 703-710.
Zhao R. and Houry W. A. (2007).
Molecular interaction network of the Hsp90 chaperone system. Adv Exp Med Biol 594, 27-36.