Биологические
науки/6.Микробиология
Квач С.В.1, Ерошевская
Л.А.1, Шахбазов А.В.2, Картель Н.А.2, Зинченко
А.И.1
1-Институт микробиологии НАН Беларуси, Минск
2-Институт генетики и цитологии НАН
Беларуси, Минск
ПОЛУЧЕНИЕ И ОЧИСТКА
РЕКОМБИНАНТНОЙ УРИДИНФОСФОРИЛАЗЫ ESCHERICHIA COLI
Как
известно, ключевой стадией синтеза модифицированных нуклеозидов является
присоединение гетероциклического основания к активированному остатку сахара –
гликозилирование. Часто эту стадию осуществляют с помощью ферментов – нуклеозидфосфорилаз
[1–3]. Нуклеозидфосфорилазы E. coli представлены
тремя внутриклеточными ферментами: уридин-фосфорилазой (УРФазой) и
тимидинфосфорилазой, использующими в качестве субстратов пиримидиновые
нуклеозиды, и пуриннуклеозид-фосфорилазой, субстратами которой являются пуриновые
нуклеозиды.
Применение стандартных методов очистки (осаждение сульфатом аммония,
хроматография на ионообменных смолах, гель-проникающая хроматография и т.д.)
достаточно трудоемки и приводят к высоким потерям целевых белков [4]. В тоже
время рекомбинантные технологии дают возможность получать химерные белки,
несущих на С- или N-концах
дополнительные пептиды, позволяющие
использовать для очистки рекомбинантных белков аффинную хроматографию. Среди
таких пептидов наибольшее распространение получил полигистидиновый олигопептид,
состоящий из 6–10 остатков гистидина и
обеспечивающий аффинность к ионам двухвалентных металлов (Ni2+, Cu2+, Co2+).
Целью данной
работы было создание штамма-суперпродуцента рекомбинантной УРФазы, несущей на
С-конце полигистидиновый олигопептид и ее очистка.
Материалы и методы. Ген udp, кодирующий УРФазу, был
выделен методом ПЦР с использованием в качестве матрицы геномной ДНК штамма E. coli
БМТ-4Д/1а [5]. Для амплификации гена udp
использовали следующую пару праймеров: UDP-F
(5′-CGTAAGCATATGTCCAAGTCTGATGTTTTT-CATCTCG-3′) и UDP-R (5′-ACTCGAGCAGCAGACGACG-CGCCGC-3′). В 5′-окончания
праймеров были встроены сайты рестрикции (выделены курсивом) эндонуклеаз NdeI (UDP-F) и
XhoI (UDP-R). Для проведения реакции амплификации использовали 1
ед. активности Fusion HF-полимеразы (New England
Biolab, США). Синтезированный продукт выделяли из геля,
обрабатывали смесью рестриктаз NdeI и XhoI, и полученную вставку с геном udp лигировали в вектор pET24b+,
содержащий T7-промотор и полигистидиновый олигопептид на С-конце (Novagen,
США). В результате была сконструирована плазмида pETUDP17. Этим вектором
трансформировали клетки E. coli BL21(DE3)
с получением штамма, обозначенного E. coli pUDP17.
Клетки E. coli pUDP17 выращивали на LB-среде, содержащей 50
мкг/мл канамицина, до оптической плотности 0,6 (λ = 600 нм); затем
проводили индукцию синтеза белка путем внесения изопропил-β-D-тиогалактопиранозида
(IPTG) до концентрации 1 мМ и продолжали культивирование в
течение 3 ч. По окончании культивирования клетки осаждали центрифугированием в
течение 10 мин при 5000 g. Осадок клеток ресуспендировали
в лизирующем буфере, содержащем 50 мМ NaH2PO4, 300 мМ
NaCl и 20 мМ имидазол (рН 8,0). Ультразвуковую дезинтеграцию клеток
осуществляли, используя режимы: мощность 0,5 кВт, температура 4оС, продолжительность
5 мин.
После
центрифугирования образца при 20000 g в течение 30 мин супернатант наносили на
хроматографическую колонку со смолой Ni-NTA (“Qiagen”, США). Колонку промывали
буфером, содержащим 20 мМ Трис-HCl, 0,5 М NaCl и 20 мМ имидазол (рН 7,9). Белок
элюировали буфером «B», содержащим 20 мМ Трис-HCl, 0,5 М NaCl и 1 М имидазол
(рН 7,9). Раствор белка подвергали диализу против 1000-кратного объема 10 мМ К-фосфатного буфера (рН 6,4), содержащего 1 мМ EDTA и
2 мМ β-меркаптоэтанол.
Активность УРФазы
определяли спектрофотометрически по изменению поглощения при распаде уридина до
урацила (λ=290 нм) [3]. За единицу активности УРФазы принимали такое её
количество, которое обеспечивало образование 1 мкмоль продукта за 1 мин в
условиях проведения реакции.
Результаты и обсуждение. В настоящей работе создана
конструкция, состоящая из гена udp,
встроенного в вектор pET24b+. В результате ген УРФазы поставлен под контроль
Т7-промотора, а на С-конце имеет последовательность, кодирующую
шестигистидиновый олигопептид. Этой плазмидой трансформировали клетки E. coli BL21(DE3), предназначенные для
экспрессии белка, находящегося под контролем T7-промотора.
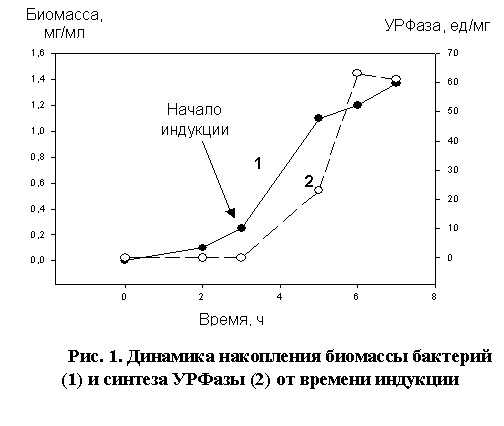
У полученного
штамма (E. coli pUD17) была проведена
индукция синтеза УРФазы в рекомендуемых условиях с использованием IPTG в
концентрации 1 мМ. Результаты этого эксперимента представлены на рис. 1.
Из рис. 1
видно, что максимальная активность УРФазы (63 ед/мг) наблюдается при индукции в
течение 3-х час. При дальнейшем культивировании активность УРФазы незначительно
падает (до 61 ед/мг на 4-й час индукции). Таким образом, оптимальными условиями
культивирования для получения максимальной активности УРФазы в штамме E. coli pUDP17 являются:
культивирование бактерий при 37oС в течение 3-х час
после индукции 1 мМ IPTG.
Далее, проводили препаративное выращивание клеток штамма E. coli pUDP17 и очистку УРФазы с
использованием металлоаффинной хроматографии. Результаты эксперимента суммированы
в табл. 1.
Таблица 1
Выделение и очистка УРФазы
|
Стадия |
Общая активность, ед |
Общий белок, мг |
Удельная активность ед/мг |
Степень очистки |
Выход, % |
|
1. Ультразвуковая
дезинтеграция клеток |
4 025 |
35 |
115 |
1 |
100 |
|
2. Выделение на смоле Ni-NTA
и диализ |
3 410 |
16 |
213 |
1,9 |
84,8 |
Визуально чистоту полученного ферментного препарата контролировали с
помощью полиакриламидного SDS-электрофореза (рис. 2).
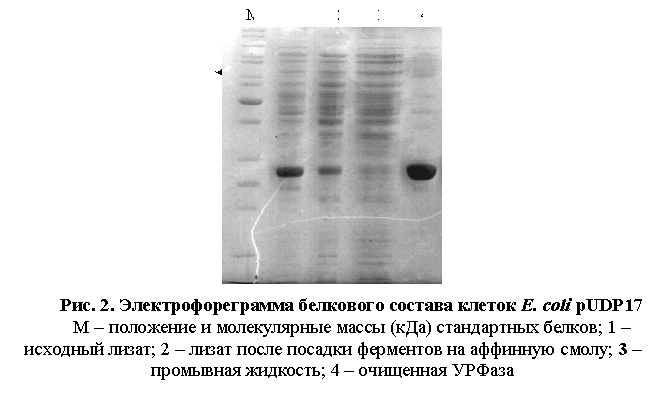
Из электрофореграммы видно, что в исходном лизате основную массу наработанного
белка составляет целевой белок. Однако при очистке белка методом
металлоаффинной хроматографии некоторое количество УРФазы не связалось со смолой,
что может быть связано с недостатком емкости сорбента. После диализа получен
белок с высокой степенью чистоты.
Из табл. 1 видно, что в результате использованного нами подхода получен
препарат фермента УРФазы, очищенной в 1,9 раза с сохранением 84,8% исходной
активности. Данный ферментнай препарат может быть использован для постановки
реакций трансгликозилирования с целью получения широкого спектра
фармакологически важных нуклеозидов.
Литература:
1. Mikhailopulo, I.A. Biotechnology
of nucleic acid constituents – state of the art and perspectives / I.A. Mikhailopulo // Curr. Org. Chem.– 2007. – Vol. 11, № 4. –Р. 317–335.
2. Immobilized biocatalysts for the production of
nucleosides and nucleoside analogues by enzymatic transglycosylation reactions
/ G. Zuffi [et al.] // Biocatal. Biotransform. –
2004. – Vol. 22. – № 1. – P. 25–33.
3. Krenitsky, T.A. Purine nucleoside
synthesis, an efficient method employing nucleoside phosphorylase / T.A.
Krenitsky, G.W. Koszalka, J.V. Tuttle // Biochemistry. – 1981. – Vol. 20, № 12. – P. 3615–3621.
4. Overexpression of Escherichia coli genes encoding nucleoside phosphorylases in the
pET/BL21(DES) system yields active recombinant enzymes / R.S. Esipov [et al.]
// Protein Expr. Purif. –2002. –Vol. 24, № 1. –P. 56–60.
5. An universal biocatalyst for the preparation of
base- and sugar-modified nucleosides via
an enzymatic transglycosylation / V.N. Barai [et al.] // Helvetica Chim. Acta. –2002. –Vol. 85, № 7. –Р. 1901–1908.