Федоренко К. В., Литвин В. А.,
Галаган Р. Л.
Черкаський
національний університет імені Богдана Хмельницького
ОДЕРЖАННЯ,
ФРАКЦІОНУВАННЯ ТА ДОСЛІДЖЕННЯ КИСЛОТНО-ОСНОВНИХ ВЛАСТИВОСТЕЙ СИНТЕТИЧНИХ
АНАЛОГІВ ФУЛЬВОКИСЛОТ
Продукти окиснювальної конденсації багатоатомних фенолів та
їх по-хідних мають велику подібність до фульвокислот (ФК) – природних
про-дуктів деструкції лігніну, які входять до складу ґрунтового гумусу.
Синтетичні аналоги цих речовин одержують і використовують з лікуваль-ною метою
у медицині та ветеринарії починаючи з початку минулого століття, коли
у Німеччині було запатентовано перший з таких процесів [1-4].
На наш погляд існує щонайменше три аргументи на користь
розвитку таких досліджень:
·
хімічний синтез дозволить краще зрозуміти природні процеси;
·
існує проблема створення стандартних зразків гумінових речовин
певного складу і властивостей;
·
існує проблема стандартизації методів дослідження елементного
складу природних гумінових
речовин.
Одним з давно відомих шляхів одержання синтетичних ФК є
окиснювальна конденсація хінонів або фенолів. Так, автором [5] синтезова-но і
охарактеризовано велику кількість таких продуктів, попередниками яких були
фенольні сполуки найрізноманітнішої будови. Було досліджено їх фізіологічну
активність і показано, що більшості з них, як і природним фульвокислотам,
притаманна чітко виражена бактеріостатична дія та здат-ність стимулювати
регенеративні процеси у тканинах вищих організмів.
В нашій роботі як
попередник синтетичних фульвокислот було використано природний барвник
гематоксилін (7,11b-дигідро-цис-(+)-бенз-(b)індено[1,2-b]піран-3,4,6а,9,10(6Н)-пентол).
Синтез проводили в установці зображеній на
рис. 1. Гематоксилін моногідрат масою 3.203 г (10 ммоль),
зважений у поліетиленовому бюксі, вміщували у колбу К’єльдаля місткістю 1 дм3. Обережно,
по стінці, приливали розчин, що містить 3.2 г NaOH (80 ммоль) та
дистильовану воду, до загального об’єму 100 см3. Повітря з
системи витискували слабким потоком
чистого електролітичного кисню. Після герметизації апаратури вмикали механічне
струшування колби-реактора, внаслідок якого відбувалось змішування реагентів,
розчинення наважки гемато-ксиліну і розпочинався процес окиснення.
1. урівнювалььна посудина 2. вимірювальний циліндр 3. колба-реактор 4. штуцер для напуску
кисню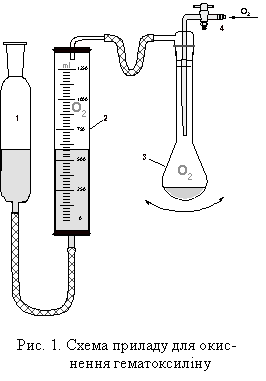
Процес вважали завершеним, коли
споживання кисню знижувалося до рівня меншого за 0.01 моль О2, з
розрахунку на 1 моль гематоксиліну за одну годину. Після завершення окиснення
реакційну суміш пропускали через колонку, заповнену катіонітом КУ-2-8 у
Н-формі. Відбір елюату починали при
появі коричневого забарвлення розчину, який витікає з колонки. Відбір елюату
припиняли, як тільки його колір змі-нювався з темно-коричневого на
світло-червоний. Об’єм одержаної при цьому фракції практично дорівнював об’єму
введеної в колонку реакційної суміші. У процесі фільтрації спостерігалось також
виділення СО2, тому, для
повного видалення карбон(IV) оксиду, зібраний
елюат нагрівали до кипіння.
Для розділення одержаної суміші на
окремі фракції нами було використано особливості
взаємодії її компонентів з барій гідроксидом Ba(OH)2.
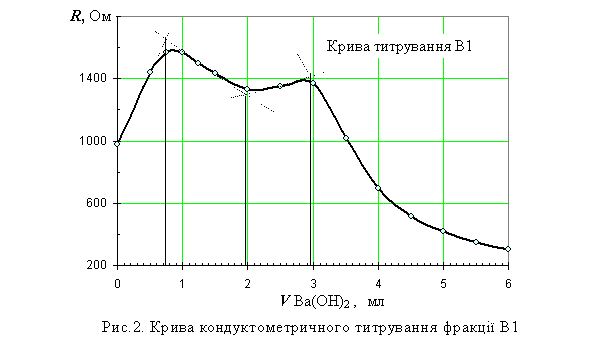
Крива алкаліметричного кондуктометричного
титрування (рис.2) суми фракцій, яку ми позначаємо як
В1, має три
перегини, що відповідають, на нашу думку, осадженню трьох субфракцій з істотно
різною спорідненістю до Ва2+-йона. Приймаючи таку гіпотезу, можемо
розрахувати об’єм осаджувача Ва(ОН)2, необхідний для препаративного
осадження першої субфракції. Осад барієвої солі відфільтровували під вакуумом
через баритовий фільтр, промивали водою і разом з фільтром переносили в стакан,
що містив розраховану кількість водної суспензії катіоніту КУ-2 в Н-формі.
Через 5-6 годин осад барієвої солі розчинявся внаслідок реакції обміну з
катіонітом.
Відфільтрувавши катіоніт, одержували першу
субфракцію, яка має кодове позначення В1\1. До фільтрату, що залишився після відділення першого осаду
барієвої солі, знову додавали розрахований за кривою титрування об’єм
осаджувача і описану процедуру повторювали. Криві титрування одержаних субфракцій у свою чергу мали більш
ніж один перегин, що в рамках прийнятої гіпотези вказує на їх неоднорідність, а
значить, можливим є подальший поділ на фракції.
Рис.3. Схема фракціонування
синтетичних фульвокислот
У нашій роботі ми не ставили за мету провести
глибоке фракціону-вання, а лише продемонструвати продуктивність такого підходу.
Всього було реалізовано два рівні
фракціонування, показані на схемі (рис.3).
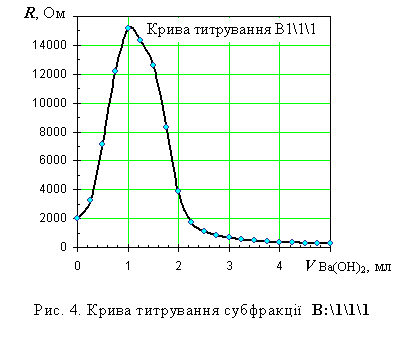
На рис.4 наведено криву титрування
субфракції В1\1\1 розчином барій гідроксиду. Видно, що тепер крива має лише
один перегин, а отже дана субфракція об’єднує молекули ФК з близькими
властивостями. Привертає також увагу високе значення електричного опору системи
у точці еквівалентності, що вказує на доволі низьке значення добутку
розчинності барієвих солей ФК з цієї субфракції.
Кислотно-основні властивості
поліелектролітів, якими є природні та синтетичні ФК, зручно характеризувати
кривими розподілу молярної част-ки протоногенних груп q за величиною їх
рК, так званим рК-спектром [6].
(1) ,
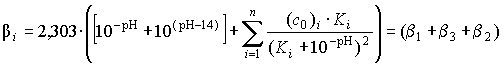
За фізичним змістом величина q пропорційна буферній ємності b системи у
будь-якій точці кривої титрування, і, отже, може виражатися загальним рівнянням
буферної ємності [7]
яке одержується комбінуванням виразів для Кдис. протоліту,
йонного добутку води, електронейтральності системи та збереження кількості
речовини протоліту. Це рівняння містить суму трьох b-функцій
від концентрації Н+. Розподіл
за рК іоногенних груп досліджуваного протоліту можна знайти застосувавши
варіаційний принцип при заданому кроці зміни К. Ця модель є надзвичайно
зручною для теоретичного дослідження поведінки складних протолітичних систем.
Нами
запропоновано алгоритм обчислення цього розподілу за даними потенціометричного
титрування. Значення рН у ході титрування з автоматич-ного титратора через АЦП надходять у
комп’ютер і зберігаються у текстовому файлі, який може опрацьовуватись
спеціально розробленим MathCAD-доку-ментом. Для побудови рК-
спектру використовується функція b2(рН), де b2 = А·![]() . Нормуючий
. Нормуючий  множник А визначається з
умови
множник А визначається з
умови
де рН1 і рН2 – точки
мінімуму функції b2 відповідно з лівого та правого краю області
зміни рН.
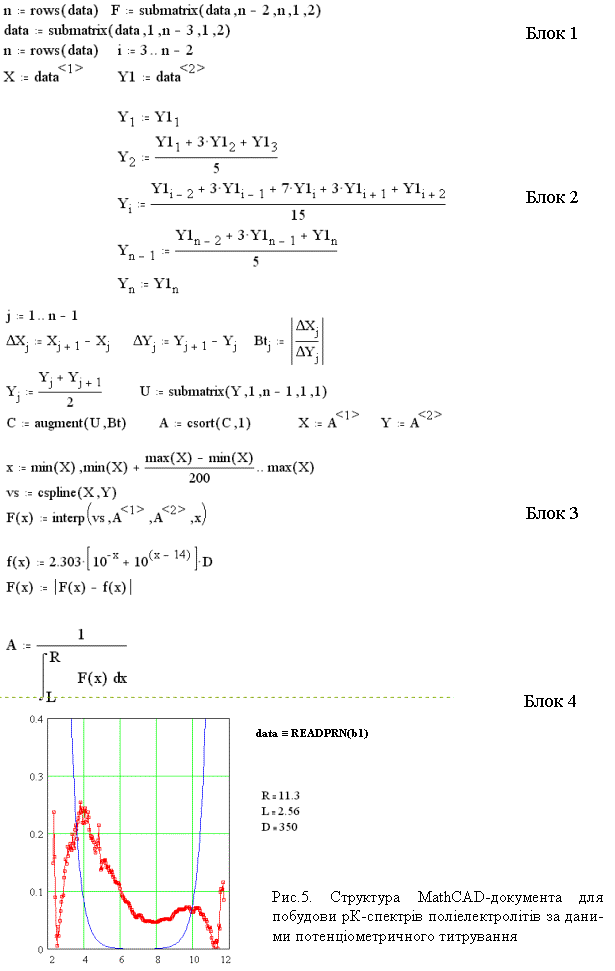
MathCAD-документ, призначений для реалізації
описаного алгоритму показано
на рис. 5. Структурно він складається з чотирьох блоків.
Блок 1 – це початкові установки та
читання даних з файла: у змінній Х зберігається об’єм титранту, а у змінній Y1 – відповідні значення рН.
Вихідні дані зазвичай потребують
згладжування, яке в даному документі
виконується за методом зваженого середнього (блок 2).
Блок 3 – це наближене обчислення
значення буферної ємності в кожній точці кривої титрування, як відношення
скінченних різниць DV/DpH. Одержані значення апроксимуються
кубічним сплайном, а значення буферної ємності в області високих і низьких
значень рН розраховуються теоретично за рівнянням
(3)
![]() .
.
Блок 4 – обчислення нормуючого
множника та побудова графіка розподілу буферної ємності системи від рК, який і
є шуканим рК-спектром.
 Контроль роботи алгоритму здійснювали шляхом одержання
рК-спектрів низькомолекулярних органічних електролітів, таких як алізарин чи
8-оксихіно-лін, та зіставлення максимумів розподілу з літературними даними по
констант-тах дисоціації і протонування. На рис. 6 наведено рК-спектри
сумарного препарату В1 синтетичних ФК та 8-оксихіноліну.
Контроль роботи алгоритму здійснювали шляхом одержання
рК-спектрів низькомолекулярних органічних електролітів, таких як алізарин чи
8-оксихіно-лін, та зіставлення максимумів розподілу з літературними даними по
констант-тах дисоціації і протонування. На рис. 6 наведено рК-спектри
сумарного препарату В1 синтетичних ФК та 8-оксихіноліну.
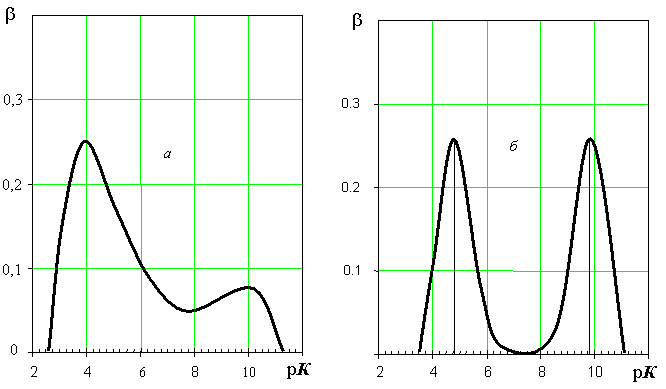
Рис.6.
рК-спектри синтетичних ФК, одержаних окисненням гематоксиліну (а)
і
низькомолекулярного органічного амфоліту 8-оксихіноліну (б)
Взаємодія солі 8-оксихіноліну з лугом виражається
рівняннями:
[С10Н7NH(ОН)]+ Сl–
+ NaOH ® [C10H7N(OH)]0¯ + NaCl + H2O
І ІІ
[C10H7N(OH)] + NaOH ® Na+ [C10H7NO] – + H2O .
ІІ ІІІ
Довідник [8] дає для константи дисоціації онієвого
йона І значення рК рівне 4,8, а для константи дисоціації гідроксигрупи у
нейтральній молекулі ІІ з утворенням аніона ІІІ, значення рК рівне 9,8.
Як видно з рисунка 2.11
максимуми розподілу за рК для 8-оксихіноліну лежать саме при цих значеннях, що
свідчить про правильність роботи розрахункового алгоритму.
Крива розподілу протоногенних
груп для досліджуваного препарату синтетичних фульвокислот характеризується
двома максимумами при рК = 4 і рК = 10. Такий характер розподілу притаманний
також природним фульвокислотам, і, згідно з загальноприйнятою концепцією [9], перший максимум приписують «карбоксильній», а другий – «фенольній» кислотності.
Література
1.Welte E., Schatz M., Ziechmann W.
Über Synthesehuminsäuren (Oxydationsmessungen). // Naturwissenschaften,
1910, 41, 213.
2.Eller W. Studien über
Huminsäuren. 4. Darstellung und Eigenschaftten künstlicher und
natürlicher Huminsäuren. // Liebigs Ann. Chem., 1923, 431,
133.
3.Seubert
B., Beilharz H., Fickert W.,
Jeromin G., Spitaler U. Method for the preparation of low molecular
weight alkali metal huminates. Pat.DE3707910, 1988.
4. Seubert B., Riede U. Process for producing huminates. Pat.ЕР0537427, 1993.
5.Laub R. J. Process for preparing synthetic soil-extract material and medicaments based thereon. US Patent № 5945446, Aug. 31, 1999.
6.Гармаш А. В., Устинова И. В., Кудрявцев А. В., Воробьева О. Н. и др. Потенциометрический анализ сложных протолитических систем методом рК-спектроскопии с использованием линейной регрессии. // ЖАХ, 1998, Т.53, № 3, С.241-248.
7.Блументаль Г., Энгельс З., Фиц И., Хабердитцл В.//Анорганикум. Под. ред. Л. Кольдица. – М.: Мир, 1984. – С. 236-243., - 653с.
8.Лурье Ю. Ю. Справочник по аналитической
химии: Справ. изд. – 6-е изд., перераб. и доп. – М.: Химия, 1989. –
448с.
9.Перминова И. В. Анализ, классификация и прогноз свойств гумусовых кислот.
Дисертация на соиск. уч. степ. докт. хим. наук, –М:. МГУ, 2000, -с.319.