Химия и
химические технологии/6. Органическая химия
Аракелян А.С., Болян Л. А.,
Джанджулян Ж. Л., Геворкян А. А.
Инверсия зависимости
нуклеофугности и реакционной
способности атомов молекул
Центр органической и фармхимии НАН
Армении, Армения.
Считается, что
реакционная способность молекул резко
падает как в гомологическом ряду,
так и при замене хорошего нуклеофуга на плохие нуклеофуги (C-J<<C-Br<C-Cl<<<C-F) [1,2]. По существу, реакционная
способность молекул падает параллельно росту величин зарядов атомов связи
С-нуклеофуг: 0.11>0.41>0.61> >1.43 (все в единицах р – полинга
[3]). Вопреки сказанному, нами обнаружен факт, согласно которому в ряду молекул
2 (схема 1) обнаруживается закономерность, противоположная ожидаемой по теории химии
[1,2]. Это при условии, что катализ почти одинаково происходит как под влиянием
молекул CuCl, так и CuBr: MeOCH2NEt2<EtOCH2NEt2< <BuOCH2NEt2 < iPrOCH2NEt2.
Причем в качестве признака роста нуклеофугности принят
выход основания Манниха. В этой связи спрашивается: может ли классическая
теория химии понять причину этой инверсии реакционной способности или
предвидеть её? Естественно, - нет; она не может (просто не в состоянии) решить
ни одну из этих задач, поскольку для этой теории медь обеих молекул – одновалентный атом металла.

Это при условии, что по зарядной силе молекулы разнятся друг от
друга именно по приведенной градации. Вопреки “классической” (и общепринятой)
теории, зарядная теория такие проблемы решает в течение считанных минут в
единицах заряда. Оценка (расчет) зарядов довольно подробно описана в наших
публикациях [3-7]. Такие же оценки (расчеты) зарядов атомов в полингах для
молекул 2a-d мы демонстрируем на Схеме 2.
 Это различие обусловлено тем, что в
общепринятой теории сродство атомов молекул оценивают не в единицах заряда, а в
единицах энергии или скорости и допускают, что реакционные способности молекул R-X (где Х – нуклеофуг) растут
согласно ряду 1о<2<о3о. Но, вопреки
этому обобщению, дибромиды 1-алкенов (всегда
и в основном) приводят к
образованию 2-бром-1-алкенов. Аналогично (тоже всегда) первичные бромиды дают высокие выходы эфиров спиртов с выходами, резко
падающими в том же ряду: 1о>>2о >>3о.
В этом отношении более показательны данные реакции замещения тетрахлорида 6
[8], при котором единственным продуктом становится не монофторид 7, а трифторид
8. Видимо, суперкислота Ола SbF5, наделенная зарядом величины +9.65
р, сначала атакует атом фтора молекулы HF и высвобождает её атом водорода с непомерно малым ионным радиусом 10-24
пм [2,9] и большой способностью преодолевать противодействие атома углерода
(Схема 3). Таким образом он начинает и завершает реакцию именно у этого a-атома углерода по принципу ЖМКО
Пирсона [10]. По той жа схеме, но взамен SbF5, роли суперкислот
играют атомы меди молекул CuBr и CuCl.
Это различие обусловлено тем, что в
общепринятой теории сродство атомов молекул оценивают не в единицах заряда, а в
единицах энергии или скорости и допускают, что реакционные способности молекул R-X (где Х – нуклеофуг) растут
согласно ряду 1о<2<о3о. Но, вопреки
этому обобщению, дибромиды 1-алкенов (всегда
и в основном) приводят к
образованию 2-бром-1-алкенов. Аналогично (тоже всегда) первичные бромиды дают высокие выходы эфиров спиртов с выходами, резко
падающими в том же ряду: 1о>>2о >>3о.
В этом отношении более показательны данные реакции замещения тетрахлорида 6
[8], при котором единственным продуктом становится не монофторид 7, а трифторид
8. Видимо, суперкислота Ола SbF5, наделенная зарядом величины +9.65
р, сначала атакует атом фтора молекулы HF и высвобождает её атом водорода с непомерно малым ионным радиусом 10-24
пм [2,9] и большой способностью преодолевать противодействие атома углерода
(Схема 3). Таким образом он начинает и завершает реакцию именно у этого a-атома углерода по принципу ЖМКО
Пирсона [10]. По той жа схеме, но взамен SbF5, роли суперкислот
играют атомы меди молекул CuBr и CuCl.
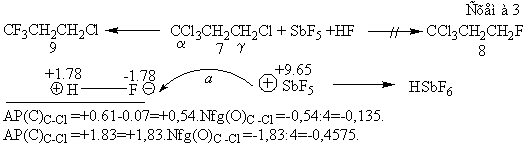
Разгадка “аномального
поведения нуклеофугов” молекул 2a-d тоже имеет свою зарядную
причину.
 Она становится очевидна, если сопоставим
соотношения продуктов отщепления по правилам Зайцева и Гофмана (Табл. 1) [1]:
чем больше заряд противоиона
катионоидной частицы, тем больше склонность катионоидной частицы атаковать
анионный центр связи С-Н. Именно по этой причине четвертичный аммониевый атом
азота, имеющий заряд порядка -1.96 р, (а в бромидах и йодидах даже -2.04 и - 2.34 р, соответственно) образует больше продукта отщепления по Гофману, чем
даже фториды связи C-F [11]. Это значит, что по мере атаки электрофила на
анионный центр аммониевого азота [2-7] происходит зарождение катионоидного
атома углерода, который атакует именно
углерод связи С-Н, носящий больший отрицательный заряд. Теперь обратим
внимание на величины положительных зарядов метиленовых атомов углерода молекул
2a-d (Схема 2). Они соответствуют величинам отрицательных зарядов нуклеофугов
тех же молекул. Сопоставляя нуклеофугности трихлор- и хлорметильной групп тетрахлорида 6 с их реакционными
способностями (Схема 3), можно обнаружить непомерно большую разницу в величинах
зарядов нуклеофугности этих фрагментов молекулы 7. Значит, фрагменты молекулы 7
реагируют по принципу ЖМКО Пирсона, обнаруживая “аномальное” замещение. Оно
тоже имеет строгую зарядную причину. Это то, что кислота Ола HSbF6 обнаруживает суперкислотность лишь потому, что
взаимодействие SbF5 с H-F происходит как атака
суперкислоты (SbF5) Ола с величиной заряда +9.65 р,
способной взаимодействовать с атомом фтора молекулы HF (основанием Полинга с зарядом -1.78 р).
В итоге высвобождается протон с зарядом +1.78 р, наделенный
суперкислотными свойствами (Схема 4). Из схемы следует, что при наличии
суперкислоты, реакция начинается и завершается у атома связи C-нуклеофуг,
имеющей больший заряд (в данном случае -0.457 против -0.135 р). Из этого факта
следует, что положительные заряды атомов меди молекул CuBr и CuCl обнаруживают суперкислотные
свойства по отношению к эфирному кислороду молекул 2a-d. Но, как показано [2], согласно
зарядной теории, при окислении и восстановлении (О-В) происходит не окисление
одного атома другим, а перераспределение зарядов атомов реагентов и субстратов.
Именно поэтому обнаружен принцип, который гласит: 1. любая химическая реакция
инициируется атомом, у которого электрофильная сила больше. 2. в перераспределении
положительных зарядов участвуют как атомы реагента, так и субстрата. 3.
перераспределение зарядов атомов происходит только по принципам взаимного
сродства атомов Полинга. 4. версия общепринятой теории химии, согласно которой
не может существовать реакция окисления-восстановления, в которой окислитель
мог бы повысить свою степень окисления больше, чем сам имеет, просто ошибочна. Видимо (Схемы 1 и 4), таким
же доказательством этого феномена является как наблюдаемое перераспределение
зарядов атомов реагента и субстрата, так и рост каталитической активности обеих солей меди (CuCl и CuBr).
Она становится очевидна, если сопоставим
соотношения продуктов отщепления по правилам Зайцева и Гофмана (Табл. 1) [1]:
чем больше заряд противоиона
катионоидной частицы, тем больше склонность катионоидной частицы атаковать
анионный центр связи С-Н. Именно по этой причине четвертичный аммониевый атом
азота, имеющий заряд порядка -1.96 р, (а в бромидах и йодидах даже -2.04 и - 2.34 р, соответственно) образует больше продукта отщепления по Гофману, чем
даже фториды связи C-F [11]. Это значит, что по мере атаки электрофила на
анионный центр аммониевого азота [2-7] происходит зарождение катионоидного
атома углерода, который атакует именно
углерод связи С-Н, носящий больший отрицательный заряд. Теперь обратим
внимание на величины положительных зарядов метиленовых атомов углерода молекул
2a-d (Схема 2). Они соответствуют величинам отрицательных зарядов нуклеофугов
тех же молекул. Сопоставляя нуклеофугности трихлор- и хлорметильной групп тетрахлорида 6 с их реакционными
способностями (Схема 3), можно обнаружить непомерно большую разницу в величинах
зарядов нуклеофугности этих фрагментов молекулы 7. Значит, фрагменты молекулы 7
реагируют по принципу ЖМКО Пирсона, обнаруживая “аномальное” замещение. Оно
тоже имеет строгую зарядную причину. Это то, что кислота Ола HSbF6 обнаруживает суперкислотность лишь потому, что
взаимодействие SbF5 с H-F происходит как атака
суперкислоты (SbF5) Ола с величиной заряда +9.65 р,
способной взаимодействовать с атомом фтора молекулы HF (основанием Полинга с зарядом -1.78 р).
В итоге высвобождается протон с зарядом +1.78 р, наделенный
суперкислотными свойствами (Схема 4). Из схемы следует, что при наличии
суперкислоты, реакция начинается и завершается у атома связи C-нуклеофуг,
имеющей больший заряд (в данном случае -0.457 против -0.135 р). Из этого факта
следует, что положительные заряды атомов меди молекул CuBr и CuCl обнаруживают суперкислотные
свойства по отношению к эфирному кислороду молекул 2a-d. Но, как показано [2], согласно
зарядной теории, при окислении и восстановлении (О-В) происходит не окисление
одного атома другим, а перераспределение зарядов атомов реагентов и субстратов.
Именно поэтому обнаружен принцип, который гласит: 1. любая химическая реакция
инициируется атомом, у которого электрофильная сила больше. 2. в перераспределении
положительных зарядов участвуют как атомы реагента, так и субстрата. 3.
перераспределение зарядов атомов происходит только по принципам взаимного
сродства атомов Полинга. 4. версия общепринятой теории химии, согласно которой
не может существовать реакция окисления-восстановления, в которой окислитель
мог бы повысить свою степень окисления больше, чем сам имеет, просто ошибочна. Видимо (Схемы 1 и 4), таким
же доказательством этого феномена является как наблюдаемое перераспределение
зарядов атомов реагента и субстрата, так и рост каталитической активности обеих солей меди (CuCl и CuBr).
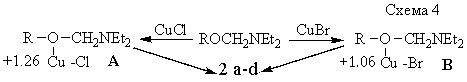
Действительно, в отсутствие этих
солей, никакая реакция не происходит. А по мере появления в реакционной среде CuCl или CuBr начинается даже экзотермическая
реакция, которая приводит к получению (очевидно, через комплексы A и B) оснований Манниха с высокими выходами. Поэтому прогрессивное
повышение выхода продукта реакции молекул 2a-d надо обусловить параллельным
повышением положительного заряда метиленового атома углерода молекул 2. Это величины зарядов стадий
уточнения d. AP(C)N-C-O (Схема 2). Справедливость
аналогии подтверждается данными [11].
Экспериментальная часть
Метилоловые эфиры 2 получили по
прописи авторов [12].
Получение оснований Манниха 3. В две колбы, снабженные обратными холодильниками,
параллельно к смеси 0.033 моля фенилацетилена и молекул 2 добавляют по 0.0075
мг CuCl и 0.01 мг CuBr. Кипятят 5 ч при
одинаковых условиях и перегонкой
выделяют амины 3, имеющие идеальные ПМР спектры.
Литература
1.
Беккер
Г. Введение в электронную теорию органических реакций. М.: Мир, 1977.
2.
Геворкян
А. А. "Обобщенная теория кислот и
оснований. Новое воззрение на реакционную способность атомов и молекул".
Изд. Наука, Ереван, 2006 г. 158 с.
3.
Геворкян
А. А., Аракелян А.С., Международная конференция
«Образование и наука XXI века», Volume 28, Praha,2012, 63-67.
4. Геворкян А. А., Аракелян А.С., Nauka I Inowscja. (Наука и инновации), Volume 14, 52-54, Poland, 2011.
5.
Геворкян
А. А. Хим. ж. Армении, 2007, 50, No 4, 713-730.
6.
Геворкян
А.А. Вестник Академии медико-технических наук РФ. 2009 No 1(2), 71-75.
7.
Геворкян А. А., Мовсисян А. А., Джанджулян Ж. Л.,
Аракелян А. С.,Торосян Г. О. Вестник
Политеха Армении. Том 2, 2010, С. 453.
8. Henne A.L., Neger N.J. J.Am.Chem.Soc.1951,73, 1042.
9.
Дж.
Хьюи, Неорганическая химия, М., «Химия», 1987, 896 с.
10. Басоло Ф. Пирсон Р., Механизмы неорганических реакций. «Мир», М. 1971.
11. Органические реакции, «Мир»,
М-1985, С.327.
12. H. Heaney, G. Papageorgeou, R. F. Wilkince. Tetrahedron, 1997, 53,
No 8, 2941-2958.