Химия и химические технологии/5.
Фундаментальные проблемы создания новых материалов и технологий
к.х.н. Алыкова Т.В., к.х.н. Салмахаева А. М.,
д.х.н. Алыков Н. М., к.т.н. Золотарева Н.В.
Астраханский государственный
университет, Россия
Квантово-химическое исследование адсорбции
тетрациклина на оксидах кремния, алюминия и алюмосиликатах
Квантово-химические
исследования системы «сорбент – сорбат» позволяют на уровне электронных
взаимодействий установить возможность направленной сорбции различных веществ.
Расчеты адсорбции молекул тетрациклина на поверхности оксидов кремния, алюминия
и алюмосиликатов выполнены с использованием полуэмпирического РМ3 метода по
программе GAMESS.
Для
реализации поставленной задачи на компьютере используется кластерный подход,
который позволяет решить проблему, связанную с оформлением и подготовкой
изучаемых моделей к квантово-химическому исследованию. Из множества элементов
поверхностей выделены схожие, тесно связанные друг с другом функциональные
группы. В результате этого сложная молекулярная модель дифференцируется на
небольшие составляющие, а все расчеты проводятся только для этих компонентов.
Модельные
кластеры поверхности кремнезема и алюмосиликата содержат неполностью координированный атом,
силанольную и силоксановую группы, адсорбированные молекулы воды, мостиковую
ОН-группу.
Граничные
условия для кластеров учитывали при помощи модели орбитально-стехиометрического
кластера (ОСК), в основу которой заложено разделение твердого тела на
ковалентно-квазинезависимые кластерные псевдоячейки. Поскольку выделяемый
фрагмент представляет собой кристаллическую псевдоячейку, то орбитальный,
электронный и остовный состав ОСК, моделирующего объемный фрагмент идеального
кристалла, должен соответствовать стехиометрии моделируемого объекта. В базисный
набор ОСК входят все валентные атомные орбитали и электроны внутренних атомов,
а от граничных атомов – только те гибридные локализованные орбитали, которые
отвечают s-связям с внутренними
атомами [1-3].
Переход
от ОСК, моделирующего объемную структуру твердого тела, к кластерным моделям
поверхностных центров, осуществляется разделением «объемного» ОСК на составные
части, соответствующие моделируемым центрам поверхности. Граничные связи
кластеров сорбентов замыкали на граничном атоме кремния Si*. Каждый
атом Si* вносит в базис ОСК одну sp3-гибридную орбиталь,
ориентированную к соседнему внутрикластерному атому кислорода.
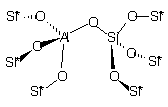
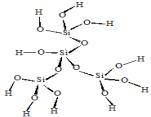
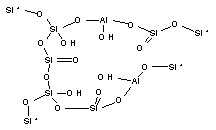
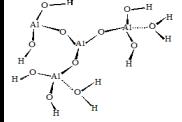
На рис. 1
приведены использованные в расчетах модели кластеров, содержащих активные
центры поверхности оксидов кремния, алюминия и алюмосиликатов (сорбент CВ-1).
|
|
|
|
|
|
кластер 1 |
кластер 2 |
кластер 3 |
|
|
|
|
||
|
кластер 4 |
кластер 5 |
||
|
Рис.1. Структуры кластеров сорбентов |
|||
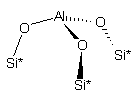
Рассмотрены свойства
и строение молекулярных моделей алюмосиликата, силикагеля (оксида кремния),
оксида алюминия и тетрациклина (ТЦ). Основными функциональными группами
тетрациклина, участвующими в образовании различных типов связей при его
адсорбции являются протонированный азот диметиламиногруппы, OH-группы, карбонильные группы
(рис. 2).
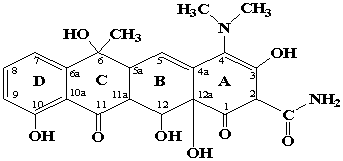
Рис. 2. Структура тетрациклина
На первоначальном
этапе, данные модели подвергались молекулярно-динамической обработке с
использованием силового поля ММ2 Элинджер [4]. На поверхности потенциальной
энергии были установлены локальные минимумы данных структур, что позволило
определить их оптимальные конфигурации.
Моделирование в
системе «тетрациклин – алюмосиликат»
Рассмотрено 38
возможных вариантов образования донорно-акцепторной и водородной связей
тетрациклина с функциональными группами алюмосиликата, на основании чего были
выделены энергетически более выгодные конфигурации взаимодействий. На рис.2
приведены лишь некоторые примеры адсорбционных систем.
|
|
|
|
|
Система 1 |
Система 2 |
Система 3 |
|
|
|
|
|
Система 4 |
Система 5
|
Система 6 |
|
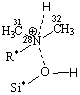
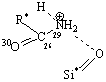
Рис. 3. Адсорбционные системы межмолекулярного взаимодействия «тетрациклин – алюмосиликат» R* -
углерод ароматического кольца |
||
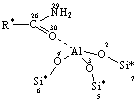
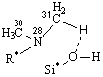
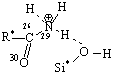
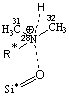
Согласно приведенным
моделям взаимодействие, преимущественно, происходит по типу донорно-акцепторной
связи Al…O и водородной – по OH- группам и диметиаминогруппе ТЦ. Смоделированные
системы представляют наибольший интерес для изучения, так как являются наиболее
устойчивыми формами существования интермедиатов.
Для описания
полученных систем использовали следующие показатели: r, Å − расстояние
между атомами реагирующих молекул; Δq, е – перенос заряда на молекулу ТЦ; DEадс, кДж/моль − энергия адсорбции. За условными
изменениями в электронной конфигурации можно проследить по значениям зарядов на
атомах (qi), определяющих некоторое
интегральное значение электронной плотности вблизи каждого атома. Об изменениях
электронной плотности приобразовании молекулярной системы из отдельных
подсистем, судят по величине переноса заряда Δq с кластера на молекулу ТЦ:
|
|
(1) |
где N – общее количество атомов, а qi – заряд i-того атома.
Энергии адсорбции
(ΔEадс, кДж/моль) формирования
предполагаемых систем рассчитывали как:
|
|
(2) |
По показателям
энергии адсорбции и длине связи между атомами, взаимодействующих молекул судят
о стабильности образующихся систем. Для наиболее выгодного положения энергия
адсорбции будет наименьшей.
В табл. 1 сведены
геометрические и энергетические параметры моделируемых систем
«тетрациклин-алюмосиликат».
Таблица №1
Геометрические и энергетические
параметры моделируемых систем
«тетрациклин - алюмосиликат»
|
АДС |
PM3 |
|||
|
r, Å |
Δq,
е |
-DEадс, кДж/моль |
||
|
1 |
O30...Al |
1,88 |
0,26 |
7,08 |
|
2 |
Si(H)O….HCH2−N28CH3 |
1,80 |
0,29 |
4,78 |
|
3 |
Si(H)O ….N28+H(CH3)2 |
2,24 |
-1,18 |
13,76 |
|
4 |
Si(H)O ….N29+(H3) |
2,64 |
-0,58 |
18,21 |
|
5 |
SiO….N28+H(CH3)2 |
2,65 |
-1,32 |
30,56 |
|
6 |
SiO….N29+(H3) |
2,69 |
-0,93 |
31,35 |
Полученные результаты
позволяют оценить энергию взаимодействия, а так же выяснить наиболее активный
центр алюмосиликата, способный легко подвергаться направленному влиянию со
стороны ТЦ.
Система 1 образуется в результате донорно-акцепторного
взаимодействия O30 карбонильной группы ТЦ с атомом Al алюмосиликата. Величины зарядов
на атомах и длины связей Al–O (1,76Å)
в алюмосиликате близки по значению с образующейся донорно-акцепторной связью
(1,87Å) это позволило
сделать предположение о формировании комплекса [AlO4]5-.
Перенос заряда на
молекулу ТЦ составляет 0,26е, это говорит о том, что происходит искажение
геометрии, а незначительное увеличение длины химической связи С26–O30 (до 1,23Å и после
1,30Å) обусловлено стабилизацией донорно-акцепторной связи O30…Al.
В системе 2 происходит образование водородной связи между водородом
метильной группы молекулы ТЦ и кислородом силанольной группы алюмосиликата.
Увеличение длины связи С–Н в метильной группе
молекулы ТЦ (до 1,10Å и после 1,12Å) свидетельствует о частичном
переносе электронной плотности от протона на кислород силанольной группы
алюмосиликата.
Система 3 образуется в результате донорно-акцепторного
взаимодействия протонированного азота (N28+) диметиламиногруппы ТЦ с атомом
кислорода силанольной группы алюмосиликата. Отрицательное значение переноса
заряда в системе (Δq= –1,18е) на протон ТЦ приводит как к геометрическому
искажению структуры в целом, так и функциональной группы, вступающей во взаимодействие,
что не наблюдается в молекулярной форме ТЦ.
Система 4 образуется в результате донорно-акцепторного
взаимодействия протонированного азота (N29+) аминогруппы ТЦ с атомом
кислорода силанольной группы алюмосиликата. Уменьшение длины химической связи N29–С26 (до 1,54Å и
после 1,50Å) и соответственно увеличение длины связи N29–Н (до 1,00Å и после
1,03Å) в ионной форме ТЦ приводит к незначительному искажению исходной
геометрии ТЦ. Однако увеличение переноса заряда (табл. 1) приводит к
противоположным выводам. Такие изменения могут быть вызваны сильной
поляризацией связи >Si–OH за счет увеличения на кислороде электронной
плотности q(–0,48)/q(–0,60) и это не наблюдается в
молекулярной форме ТЦ.
Уменьшение величины
заряда на N29 q(0,641)/q(0,472) также свидетельствует о
переносе плотности от кислорода силанольной группы кластера алюмосиликата на
протонированный азот (N29+) аминогруппы ТЦ.
Система 5 образуется в результате донорно-акцепторного
взаимодействия протонированного азота (N28+) диметиламиногруппы ТЦ с атомом
кислорода силоксановой группы алюмосиликата. Резкое увеличение переноса заряда
(Δq= –1,32e) на протон ТЦ приводит как к
геометрическому искажению по функциональной группе, так и структуры в целом.
Подобное не наблюдается в молекулярной форме ТЦ.
Система 6 также образуется в результате донорно-акцепторного
взаимодействия протонированного азота (N29+) аминогруппы ТЦ с атомом
кислорода силоксановой группы алюмосиликата. Наблюдается уменьшение длины химической
связи N29–С26
(до 1,53Å и после 1,49Å) и увеличение длины связи N29–Н (до 1,00Å и после
1,038Å) в ионной форме ТЦ, что приводит к незначительному искажению
исходной геометрии ТЦ. Однако увеличение переноса заряда (Δq = –0,93e) на протон ТЦ приводит к
противоположным выводам. Подобное не наблюдается в молекулярной форме ТЦ.
Полученные
квантово-химические результаты позволяют предположить, что в реальной системе
возможно протекание различных процессов, однако расчеты позволяют сделать вывод
о том, что наиболее устойчивыми из рассмотренных будут системы 3-6. Это системы преимущественно образованны при участи
протонированной формы молекулы ТЦ. В результате, при исследовании 38-ти моделей
АДС, можно отметить, что в среднем энергия адсорбции ТЦ на алюмосиликате
(сорбенте СВ-1) может составить около –10,50 кДж/моль.
Моделирование в
системе «тетрациклин – диоксид кремния»
Аналогичная процедура
проведена для моделирования взаимодействий в системе «тетрациклин – оксид
кремния». В результате расчетов, также были выделены энергетически более
выгодные конфигурации. Рассмотрено 14 возможных вариантов образования
донорно-акцепторных и водородных связей с ТЦ. Расчеты показали, что в среднем
энергия адсорбции ТЦ на оксиде кремния (силикагель КСМГ) может составить около
–9,35 кДж/моль.
Моделирование в
системе «тетрациклин – оксид алюминия»
Для реакционных
центров оксида алюминия, рассмотрено 26 возможных вариантов образования
донорно-акцепторных и водородных связей с ТЦ. Полученные результаты всех
модельных систем позволили предположить, что реальная энергия адсорбции ТЦ на
оксиде алюминия может составить в среднем
–8,5 кДж/моль.
Также были проведены
кспериментальные исследования сорбции тетрациклина на оксидах кремния, алюминия
и алюмосиликатах.
Сравнение
экспериментальных и теоретических данных по адсорбции ТЦ на кластерах 1 – 5
приведено в табл. 2.
Таблица №2
Энергии адсорбции ТЦ, полученные
в результате квантово-химических расчетов (ΔЕрасч) и
эксперимента (ΔЕэксп) (кДж/моль).
Расчеты проведены методом РМ3
|
Сорбат |
–
DЕрасч для кластеров |
||||||
|
КЛ-1 |
КЛ-2 |
КЛ-3 |
КЛ-4 |
КЛ-5 |
|||
|
Тетрациклин |
8,15 |
8,20 |
9,35 |
10,50 |
8,50 |
||
|
–
DЕэкс для сорбентов |
|||||||
|
Оксид
алюминия |
Силикагель
КСМГ |
CВ-1 |
|||||
|
8,41 |
9,40 |
10,47 |
|||||
Анализ
экспериментального и теоретического изучения адсорбции тетрациклина,
содержащего целый набор различных функциональных групп, позволяет сделать заключение,
сводящееся к следующему. Кластеры 1 – 5 построены идентично, все они содержат
активные центры, такие как группы:
![]() ,
,![]() ,
, ![]() ,
, ![]() ,
,![]() ,
, ![]() ,
, ![]() ,
,
т.е. у тетрациклина имеется
широкая возможность к адсорбции по различным механизмам.
Как видно из
результатов, представленных в табл. 2 экспериментальная энергия сорбции
тетрациклина на оксиде алюминия близка к теоретически рассчитанным величинам
энергии сорбции тетрациклина на кластере 5, на силикагеле КСМГ – на кластере 3
и на сорбенте СВ-1 – на кластере 4.
При оптимальном рН
сорбции ТЦ на оксиде алюминия у сорбента формируются кластеры содержащие
гидроксильные группы. Это означает, что в области максимальной сорбции на
оксиде алюминия между сорбентом и сорбатом возникают водородные связи и возникает достаточно сильное электростатическое
взаимодействие, при этом также наблюдается взаимодействие Ван-дер-Ваальсовых
сил.
На силикагеле КСМГ
максимальная сорбция тетрациклина наблюдается в слабокислой среде. В слабокислой
среде силикагель имеет кластер аналогичный кластеру оксида алюминия в
нейтральной и слабощелочной среде, поэтому в области максимальной сорбции для
силикагеля характерны электростатические взаимодействия, формирование
водородных связей, а также Ван-дер-Ваальсовые взаимодействия.
Сорбент CВ-1 является сорбентом
универсальным. Адсорбция происходит от слабокислой до щелочной среды. Сорбент CВ-1 содержит как в кислой, так и
слабощелочной среде, за счёт оксидов алюминия и кремния группы характерные
отдельно для оксида кремния и отдельно для оксида алюминия. В связи с этим
адсорбция тетрациклина на сорбенте СВ-1 протекает с формированием водородных
связей за счёт Ван-дер-Ваальсовых сил и электростатического взаимодействия в
области рН адсорбции и оксида кремния и оксида алюминия (рН 3 – 9).
Так как опоки,
используемые для производства сорбента CВ-1 содержат в своей структуре
также и кластеры, представляющие собой особое образование из силанольных и силоксановых
групп, которые являются аналогами известных в органической химии
макроциклических соединений, имеющих внутримолекулярную полость для связывания
ионов и молекул, т.е. представляющие собой неорганические ионофоры, то CВ-1 избирательно поглощает ионы
металлов, которые имеют вакантные орбитали, а так же органические соединения
содержащие первичную, вторичную и четвертичную аминогруппы. Такие группы атомов
характерны для тетрациклиновых антибиотиков.
Таким образом,
адсорбция тетрациклина сорбентом CВ-1 связана с участием в сорбционном процессе
акцептора электронных пар – положительно заряженного азота и доноров
электронных пар – кислорода силанольных, силоксановых и мостиковых групп
кластеров сорбента СВ-1. Одновременно адсорбция ТЦ сопровождается образованием
различных видов связей – водородных, Ван-дер-Ваальсовых и ионных.
Литература
1.
Алыкова,
Т.В., Алыков, Н.М., Пащенко, К.П., Воронин, И.И., Алыков, Н.Н. Моделирование
механизмов адсорбции ряда органических веществ на алюмосиликатах / Т.В.
Алыкова, Н.М. Алыков, К.П. Пащенко, И.И. Воронин, Н.Н. Алыков // Изв. вузов.
Химия и хим. технол. – 2003. – Т.46. – №6. – С. 31- 33.
2.
Алыкова,
Т.В., Пащенко, К.П. Расчёт моделей адсорбционных комплексов молекул
ароматических соединений с активными центрами поверхности кремнезёма и
алюмосиликатов / Т.В. Алыкова, К.П. Пащенко // Изв. вузов. Химия и хим. технол.
– 2004. – Т.47. – №2. – С.114-118.
3.
Алыков,
Н.М., Алыкова, Т.В., Пащенко, К.П. Квантово-химическое кластерное моделирование
адсорбции ароматических углеводородов / Н.М., Алыков, Т.В. Алыкова, К.П.
Пащенко // Естественные науки. Журн. фунд. и прикл. исслед. – 2002. – №4. – С.
206 – 214.
4.
Соловьев,
М.Е. Соловьев, М.М. Компьютерная химия./ М.Е. Соловьев, М.М. Соловьев – М.:
СОЛОН Пресс, 2005, 536 с.