НАРУШЕНИЕ ОБМЕНА ФЕНИЛАЛАНИНА
Н.В. Рылова
Казанская государственная медицинская
академия
Распространенность
Заболевание чаще встречается в северных европейских
странах – частота 1:10000, в Ирландии с частотой 1:4560, в России- 1:8-10000,
почти не встречается у негров. У здоровых
родителей, являющихся гетерозиготными носителями мутантного гена, нередко
рождаются больные дети, а родственные браки увеличивают частоту рождения
ребенка, страдающего фенилкетонурией (ФКУ). Заболевание наследуется по
аутосомно-рецессивному типу [1, 2].
Этиология и патогенез.
Открытие фенилкетонурии связывают с именем норвежского
врача Ивара
Асбьёрна Фёлинга, описавшего в 1934 году
гиперфенилаланинемию,
ассоциированную с задержкой
умственного развития. В Норвегии
заболевание известно под названием «болезни Фёлинга» в честь открывателя.
Успешное лечение впервые было разработано и проведено в Англии (Бирмингемский
детский госпиталь) группой медиков под руководством Хорста Биккеля в начале
50-х годов XX века, однако настоящий успех пришел только после широкого
применения ранней диагностики фенилкетонурии по повышенному содержанию
фенилаланина в крови у новорожденных (метод Гатри, разработанный и внедренный в
1958—1961 гг).
Со временем и накоплением опыта в диагностике и
лечении фенилкетонурии стало ясно, что это гетерогенное заболевание, вызываемое
множеством мутаций в разных генах. Выделены и описаны атипичные формы ФКУ,
разработаны новые методы лечения. В ближайшей перспективе — генотерапия
этого тяжелого заболевания, ставшего классическим образцом успешного оказания
медицинской и организационной помощи при наследственной патологии [3].
В результате мутации гена, контролирующего синтез
фенилаланингидроксилазы, развивается метаболический блок на этапе превращения
фенилаланина в тирозин, вследствие чего основным путем преобразования
фенилаланина становится дезаминирование и синтез токсических производных -
фенилпировиноградной, фенилмолочной и фенилуксусной кислот. Кроме того,
образуются также почти полностью отсутствующие в норме фенилэтиламин и ортофенилацетат, избыток которых вызывает нарушение метаболизма липидов в
головном мозге.
Окончательно механизм развития нарушений функций мозга при фенилкетонурии
остается неясным. Среди причин также предполагается дефицит нейромедиаторов
мозга, вызванный относительным снижением количества тирозина и других «больших»
аминокислот, конкурирующих с фенилаланином при переносе через
гематоэнцефалический барьер, и прямое токсическое действие фенилаланина.
Следствием нарушенного обмена в мозге является тяжелое психическое
недоразвитие. Если не предпринято своевременное лечение, то больные на всю
жизнь остаются глубокими инвалидами. В крови и тканях значительно увеличивается
содержание фенилаланина (до 0,2 г/л и более при норме 0,01-0,02 г/л).
Существенную роль в патогенезе болезни играет недостаточный синтез тирозина,
который является предшественником катехоламинов и меланина.
Отсутствие в печени фермента фенилаланиноксидазы
препятствует нормальному превращению фенилаланина пищи (рис. 1). Поэтому
фенилаланин используется лишь при синтезе белка, а избыток накапливается в
клетках печени и попадает в кровоток. Количество фенилаланина становится
токсичным для клеток мозга. Почки не справляются с его реабсорбцией, в
результате чего он выводится с мочой. Именно наличие этого фенилкетона в моче
дало основание назвать соответствующее патологическое состояние фенилкетонурией
[2, 4].
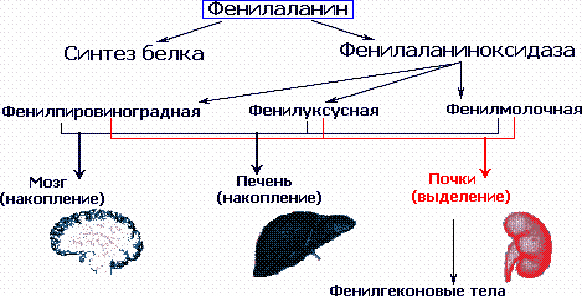
Рисунок 1. Патогенез фенилкетонурии.
Варианты ФКУ
Фенилкетонурия 1.
Классическая фенилкетонурия (ФКУ) описана
А.Folling, 1934 г.
Заболевание наследуется аутосомно-рецессивно и вызвано мутацией гена,
локализующегося в длинном плече 12 хромосомы. В основе болезни лежит дефицит
фермента фенилаланин-4-гидроксилазы. В результате метаболического блока
происходит значительное накопление в тканях и жидкостях больного организма
фенилаланина и таких его производных, как фенилпировиноградная, фенилмолочная,
фенилуксусная кислоты, фенилэтиламин, фенилацетилглютамин, и др. Частота
классической ФКУ среди новорожденных по данным массового скрининга в среднем
колеблется от 1:5000 до 1:10000 по разным регионам России [2].
В патогенезе ФКУ имеют
значение следующие механизмы:
- Прямое токсическое действие на ЦНС фенилаланина и его
производных;
- Нарушение в обмене белков, липо- и гликопротеидов;
- Расстройства транспорта аминокислот;
- Нарушение метаболизма гормонов;
- Нарушение обмена моноаминовых нейромедиаторов (катехоламинов и
серотонина);
- Нарушение функции печени - диспротеинемия, генерализованная
гипераминоацидемия, повышение ДФА, метаболический ацидоз, нарушение
окислительной и белковосинтезирующей функций клеточных органелл [1].
Фенилкетонурия 2.
Впервые атипичная ФКУ
описана I.Smith, 1974 г. Заболевание связано с дефицитом
дигидроптеридинредуктазы. Заболевание наследуется аутосомно-рецессивно. Генный
дефект локализуется в коротком плече 4 хромосомы, участке 4р15.3. В результате
недостаточности дигидроптеридинредуктазы нарушается восстановление активной
формы тетрагидробиоптерина, участвующего в качестве кофактора в
гидроксилировании фенилаланина, тирозина и триптофана. Частота заболевания
составляет 1:100000 новорожденных. Рано начатое лечение способствует
нормализации фенилаланина в крови, однако не предупреждает появление
клинической симптоматики, которая развивается в начале второго полугодия жизни.
Фенилкетонурию 2 называют диеторезистентной ФКУ.
Фенилкетонурия 3.
Этот вариант болезни описал S. Kaufman в 1978 г. Заболевание наследуется
аутосомно-рецессивно и связано с недостаточностью 6-пирувоилтетрагидроптерин
синтетазы, участвующей в процессе синтеза тетрагидробиоптерина. Развивающиеся
при этом расстройства сходны с нарушениями, наблюдаемыми при ФКУ 2. Частота
болезни составляет 1:30000 новорожденных. Фенилкетонурия 3 также
диеторезистентна.
Другие варианты ФКУ:
Эти формы ФКУ связаны с нарушением
альтернативных путей обмена фенилаланина. Формируется метилминдальная ацидурия
и парагидроскифенилуксусная ацидурия.
- Материнская фенилкетонурия.
Заболевание
развивается у потомков женщин, страдающих ФКУ и не получающих диету в зрелом
возрасте. Патогенез мало изучен, предполагается, что он сходен с патогенезом
остальных форм ФКУ. Тяжесть поражения плода коррелирует с уровнем фенилаланина
в плазме матери. Так как эмбрион особенно чувствителен к тератогенным
воздействиям, рекомендуется начинать диету еще до наступления беременности. В
суточном рационе использовать менее 15-20 мг/кг фенилаланина. При этом важно
избегать дефицита незаменимых аминокислот.
- Клиническая
фенилкетонурия.
При рождении больные фенилкетонурией не
отличаются от других новорожденных. Манифестация ФКУ происходит обычно в
возрасте 2-6 месяцев.
Клинические
проявления
Дети с фенилкетонурией рождаются без
каких-либо признаков болезни. Однако уже на втором
месяце можно заметить некоторые физические признаки: осветление волос, радужек
глаз, что особенно заметно у детей, родившихся с темными волосами. Многие дети
очень быстро и чрезмерно прибавляют в весе, однако остаются рыхлыми, вялыми. У
большинства из них рано зарастает большой родничок. Чаще всего явные признаки
болезни обнаруживаются на 4-6 месяце жизни, когда дети перестают реагировать
радостью на обращение к ним, перестают узнавать мать, не фиксируют взгляд и не
реагируют на яркие игрушки, не переворачиваются на живот, не сидят [5,6].
В течение многих лет соответствующим диагностическим
тестом служит реакция между фенилпировиноградной кислотой, которая выделяется с
мочой ребенка, и хлорным железом. При положительной реакции появляется типичное
зеленое окрашивание. Кроме того, образуются и выводятся с мочой другие
аномальные метаболиты, такие как фенилмолочная и фенилуксусная кислоты.
Последнее соединение «пахнет мышами», так что болезнь легко диагностировать по
запаху; именно так она и была впервые обнаружена [2, 3, 7].
Дети, как правило, белокурые с голубыми глазами,
светлой кожей, которая чувствительна к травматизации, у них часты проявления
экземы, дерматита; имеются папулезные высыпания (рис.2). Телосложение
диспластическое, нередко выявляются пороки сердца, уменьшение размеров черепа,
гипогенитализм и нанизм, вегетативные дисфункции, расстройства координации
движений, нарушения походки, поведения. Клинических
отличий классической и атипичных форм ФКУ не существует. Форма ФКУ выявляется только в процессе лечения. Для
атипичных форм характерно прогрессирование проявлений болезни, несмотря на
адекватное лечение с помощью диеты. При классической форме под влиянием диеты с
резким ограничением продуктов, содержащих фенилаланин, клинические проявления
редуцируются.

Рисунок 2. Внешний вид пациентки с ФКУ.
По мере прогрессирования болезни могут
наблюдаться эпилептиформные приступы - развернутые судорожные и бессудорожные
типа кивков, поклонов, вздрагиваний, кратковременных
отключений сознания. Гипертония отдельных групп мышц проявляется своеобразной
"позой портного" (поджатые ноги и согнутые руки). Могут наблюдаться
гиперкинезы, атаксия, тремор рук, иногда парезы по центральному типу.
Обнаруживается склонность к артериальной гипотензии.
При отсутствии лечения формируется задержка
статико-моторного и психоречевого развития, умственная
отсталость достигает, как правило, глубокой степени (идиотия или имбецильность,
глубокая психическая инвалидность) [6].
Дигностика фенилкетонурии
Чрезвычайно важно установить диагноз в доклинической стадии
или по крайней мере не позднее 2-го
месяца жизни, когда могут проявиться первые признаки болезни. Для этого всех
новорожденных обследуют по специальным программам скрининга (рис. 3),
выявляющего повышение концентрации фенилаланина в крови уже в первые недели
жизни.
Рисунок 3. Неонатальный скрининг на ФКУ.
В роддоме на 4-й -5-й день жизни
новорождённого(доношенного) и на 7-й день- у недоношенного берут кровь на
обследование на ФКУ. Через час после кормления каплей капиллярной крови
пропитывают специальный бумажный бланк. При концентрации фенилаланина в образце
крови более 2,2 мг% ребенка с родителями направляют в медико-генетический центр
для осмотра, дообследования, уточнения диагноза. Каждого ребенка, у которого
обнаруживаются признаки задержки развития или минимальная неврологическая
симптоматика, необходимо обследовать на патологию обмена фенилаланина.
Используют микробиологический и флюорометрический методы определения
концентрации фенилаланина в крови, а также пробу Фелинга на
фенилпировиноградную кислоту в моче (прибавление нескольких капель 5% раствора
треххлористого железа и уксусной кислоты к моче больного приводит к появлению
зеленой окраски пятна на пеленке). Эти и другие подобные методы относятся к
категории ориентировочных, поэтому при положительных результатах требуется
специальное обследование с использованием точных количественных методов
определения содержания фенилаланина в крови и моче (хроматография аминокислот,
использование аминоанализаторов и др.), которое осуществляется
централизованными биохимическими лабораториями. Дети требуют специального
наблюдения и лечения в медико-генетических центрах. Дифференциальный диагноз
проводят с внутричерепной родовой травмой, внутриутробными инфекциями [3, 7, 8,
9, 11,12].
ФКУ может быть
диагностирована на основе обнаружения следующих признаков:
- стойкой гиперфенилаланинемии (более 240 ммоль/л);
- вторичного дефицита тирозина;
- экскреции фенилкетонов с мочой (проба Феллинга на экскрецию
фенилпировиноградной кислоты).
В настоящее время, согласно приказу
Минздрава России № 316 от 30.12.93, проведение неонатального скрининга на ФКУ
стало обязательным. Скрининговые тесты должны быть простыми, недорогими и
информативными. Этим требованиям отвечают методы, используемые для ранней
диагностики ФКУ:
- микробиологический тест Гатри;
- метод флюоресцирующих антител (лабораторный комплекс
"Флюороскан", позволяющий проводить 800 проб в час);
- метод тонкослойной хроматографии.
Оптимальные сроки обследования
новорожденных - доношенных, зрелых - 5-6 день жизни; недоношенных, незрелых,
больных - 10-14 день жизни [2, 10].
Трактовка результатов:
1
группа.
Уровень
фенилаланина не превышает 200 ммоль/л (1-3 мг%) - норма;
2
группа.
Уровень фенилаланина составляет 200-500
ммоль/л (3-10 мг%) - гиперфенилаланинемия. В эту группу входят дети с
транзиторной гиперфенилаланинемией, вследствие незрелости ферментных систем
печени и больные ФКУ. За данной группой проводится наблюдение по следующему
плану: если в течение 6 недель при еженедельном исследовании уровень
фенилаланина остается менее 500 ммоль/л, контроль за уровнем фенилаланина крови
проводят до 1 года первоначально каждые 3 месяца, а затем каждые 6 мес. жизни.
При уровне более 500 ммоль/л назначается диетотерапия.
3
группа.
Уровень фенилаланина превышает 500 ммоль/л
(более 10 мг%), диагностируется ФКУ и с момента постановки диагноза назначается
диетотерапия. В настоящее время разрабатываются и внедряются
молекулярно-генетические методы диагностики генного дефекта при ФКУ. Прямая
диагностика мутантного гена проводится с помощью синтетических
олигонуклеотидных зондов, этот метод пригоден для дородовой диагностики ФКУ и
выявления гетерозиготного носительства. Помимо молекулярно-генетического
анализа, выявление гетерозигот может осуществляться биохимическими тестами
после нагрузки фенилаланином в дозе 25 мг/кг [2,7,10].
Эффективность скрининговых
тестов на фенилкетонурию.
В течение более двух десятилетий в
качестве основных скрининговых тестов на фенилкетонурию выступали
автоматизированные определения фенилаланина в крови, например, тест Гутри. Хотя
не существует каких-либо работ, в которых методически точно определялась бы
чувствительность и специфичность теста Гутри. Международный опыт использования
теста на миллионах новорожденных показал, что он выявляет практически все случаи, а имевшие место ошибки
связаны с административными или лабораторными недоработками. В качестве
альтернативного метода проверки можно использовать флюорометрические наборы,
обнаруживающие превосходную чувствительность.
Хотя в течение многих лет ложно-положительные
результаты рассматривались как менее важные, чем ложно-отрицательные, поскольку
их нетрудно скорректировать, повторив тест, все же следует иметь в виду, что
повторный анализ вызывает значительное беспокойство родителей. На
чувствительность теста Гутри влияет возраст новорожденного, когда берется
проба. Имеющаяся в настоящее время тенденция выписывать детей из родильного
дома как можно раньше (в результате чего скрининг на ФКУ выполняют в возрасте
1—2 дней от роду) заставляет ряд медиков высказать соображение, что выполненный
в таком возрасте тест может оказаться неточным.
Дело в том, что сразу после родов уровень фенилаланина
в крови обычно бывает нормальным, а затем возрастает в первые дни после
рождения. Если использовать в качестве стандарта привычный уровень 4 мг/дл, то
у ряда новорожденных с фенилкетонурей такой уровень будет достигнут только
через несколько дней. Точных данных, которые показали бы, насколько часто
встречаются подобные случаи, нет. В одном исследовании было показано, что в
течение 48 часов жизни могут быть выявлены все случаи фенилкетонурии, однако
есть сообщения, что в течение первых 24 часов число ложно-отрицательных
результатов может составить 2—6, а то и 15—16%. По данным ряда авторов,
флюорометрические наборы имеют большую чувствительность, чем тест Гутри. Было
предложено еще два решения, чтобы дополнительно повысить чувствительность —
повторять тест на всех новорожденных после выписки из клиники и понизить
пороговое значение уровня фенилаланина. Однако такой подход не разделяется
многими учеными. Повторная проверка дала бы очень незначительное число новых
обнаружений при высокой стоимости такого скрининга. Подсчитано, что для того,
чтобы обнаружить еще один дополнительный случай ФКУ надо провести от 600 тысяч
до 6 миллионов новых тестов. Если же снизить порог, то это повысит чувствительность
за счет специфичности и увеличит отношение ложно-положительных к
истинно-положительным случаям. Тем не менее во многих скрининговых программах
критический порог снижен до 2 мг/дл. В качестве средства предотвращения
осложнений у плода рекомендуется выполнять скрининг на беременных женщинах на
материнскую ФКУ. Обычно такое нарушение наблюдается редко, а большинство
женщин, больных фенилкетонурией, знают об этом. Таким образом, полезный выход
от такого скрининга очень мал. В одной программе, в которой для такого
пренатального скрининга были выполнены 260 тысяч тестов. Найдены 9 ранее
неизвестных случаев гиперфенилаланинемии. Из 11 последующих беременностей у 5
женщин была доброкачественная гиперфенилаланинемия, 4 новорожденных из этого
числа были здоровыми (пятая беременность закончилась абортом). В Массачусетсе в
течение 10 лет делался рутинный анализ крови, взятой из пуповины, за это время
были обнаружены 22 женщины с ранее недиагностированной гиперфенилаланинемией
[11,12,14].
Эффективность
ранней диагностики фенилкетонурии.
До распространения лечения пищей с ограниченным
количеством фенилаланина в начале 60-х годов, часть ФКУ приводила к тяжелым
расстройствам умственного развития детей. Обзор, выполненный в 1953 году,
показал, что у 85% коэффициент интеллекта был ниже 40, а у 37% ниже 10, только
менее 1% обнаруживали коэффициент выше 70. Большинство детей с обнаруженными
случаями фенилкетонурии оказывалось в приютах для умственно отсталых. Однако
после того, как стал известен способ получения пищи с ограниченным количеством
фенилаланина, более 95% детей с ФКУ развиваются с нормальным или близким к
нормальному интеллектом. В одной большой многолетней работе прослеживали
коэффициент интеллектуального развития у 100 детей, за которыми следили до
8-летнего возраста, он был на уровне среднего. В другом исследовании также
показано, что подростки и молодые люди, у которых при рождении была обнаружена
фенилкетонурия, вполне органично вписывались в нормальную жизнь.
Эффективность диетических ограничений фенилаланина
никогда не проверялась в тщательно построенных методических исследованиях, но и
в будущем вряд ли такое исследование будет выполнено по этическим соображениям.
Тем не менее разительный контраст между детьми, пораженными фенилкетонурией, и
теми, которых удалось вылечить с помощью диеты, побудили большинство
правительств западных стран ввести тест на ФКУ, как обязательную скрининговую
проверку [15,16,].
К сожалению, имеются ограничения и касательно
эффективности диеты. Чтобы предотвратить необратимые последствия
фенилкетонурии, необходимо ввести ограничения на фенилаланин с самого раннего
возраста, и такую диету иногда приходится выдерживать 4—8 лет. В настоящее
время появились данные, свидетельствующие о целесообразности придерживаться
такой диеты также и в подростковом возрасте, а то и взрослым. И даже если все
эти меры приняты, диета не всегда обеспечивает полную защиту от некоторых более
тонких проявлений ФКУ. Проверка интеллектуальных способностей лиц, перенесших
ФКУ, хотя и лежит в пределах нормы относительно больших масс населения, все же
значительно ниже, чем у сверстников и у родителей. Сообщают также о некоторых
психологических изъянах, таких, как: перцептуальная моторная дисфункция,
трудности обучения. Здесь помощь может оказать ранее обнаружение материнской
фенилкетонурии.
Частота материнской фенилкетонурии увеличивается с
ростом числа вылеченных в детстве от фенилкетонурии женщин, которые вступают в
детородный возраст. Материнская гиперфенилаланинемия может вызывать
тератогенные эффекты даже относительно нормального плода, не отягощенного
генетической ФКУ. Если мать во время беременности не придерживается диеты с
низким содержанием фенилаланина, то существует очень большой риск рождения
больного ребенка. Более 90% таких детей будут умственно отсталыми, у 75% будет
наличествовать микроцефалия, у 40—50% замедление роста в матке, у 10—25% -
другие дефекты. Нет уверенности, что можно предотвратить эти случаи за счет
правильной диеты с ограничением фенилаланина. В ряде исследований было
показано, что диетические ограничения не позволяют полностью устранить
поражение плода [11, 12, 14, 15].
Рутинный скрининг всех новорожденных на фенилкетонурию
обязателен во всех штатах Америки. Американская академия педиатрии рекомендует
брать пробу из пятки до того, как ребенок покидает клинику перед самым моментом
выписки. Недоношенные дети и такие, которых лечили по поводу ФКУ, должны
проверяться на 7-й день или накануне. Академия рекомендует делать повторный
тест до третьей недели жизни для детей, у которых первый тест был выполнен в
течение первых 24 часов жизни. Некоторые авторы выступают за рутинный
пренатальный скрининг материнской ФКУ, но большинство групп медиков не
поддерживают такой подход, учитывая высокую стоимость подобного скрининга. Для
всех новорожденных рекомендуется делать анализ крови на уровень фенилаланина до
выписки из роддома, предпочтительно в возрасте старше 3 дней. Для
новорожденных, которых проверяли в течение первых 24 часов, скрининговый тест
следует повторить в течение первых 14 дней жизни. Всех родителей нужно
надлежащим образом информировать относительно правильной интерпретации
результатов теста на ФКУ, упоминая также о возможности ложно-положительных
результатов. Проводить рутинный пренатальный скрининг на материнскую ФКУ не
рекомендуется.
Теперь только в США ежегодно на фенилкетонурию
проверяются более 3 млн новорожденных. Результаты диагностики показали, что
ежегодно при расходах 5—10 млн долларов выявляются только 183 случая
фенилкетонурии, и некоторые специалисты считают это расточительством. Однако
Роберт Гатри, ведущий эксперт и сторонник обязательной диагностики, утверждает:
расходы на выявление одного случая фенилкетонурии (при условии, что проводится
10 тыс. анализов, каждый стоимостью от 50 центов до 4 долларов) составляют сумму
до 50 тыс. долларов; но если не проводить диагностику, то о больном следует
заботиться всю его оставшуюся жизнь (продолжительность которой в среднем
составляет 50 лет, а ежегодные расходы на содержание в лечебном учреждении
составляют по самым скромным оценкам 5 тыс. долларов ежегодно). В эти 250 тыс.
долларов еще не входят дополнительные расходы. О том, что испытывают родители
безнадежно больных детей, и вовсе говорить не приходится. Если не обсуждать
человеческие ценности, а хотя бы задаться экономическим вопросом, что лучше —
потратить 50 тыс. долларов сейчас или в пять раз больше впоследствии? При
условии совмещения в одном анализе от одного ребенка диагностики нескольких
наследственных заболеваний экономический эффект станет еще больше.
Диеторезистентные формы ФКУ диагностируют
при помощи:
- исследования биоптеринов мочи ;
- перорального нагрузочного теста с тетрагидробоиптерином (через
4-6 часов после однократной дачи нагрузки в дозе 7,5 мг/кг массы тела
происходит резкое снижение и нормализация уровня фенилаланина в крови с
одновременным повышением уровня тирозина);
- исследования активности дигидроптеридинредуктазы и
6-пирувоилтетрагидроптерин синтетазы в культуре кожных фибробластов,
эритроцитах, гепатоцитах [7, 10].
Наследственность
Болезнь наследуется по рецессивному типу: т.е. болеют
сестры и братья из одной семьи, а родители здоровы, хотя и являются
гетерозиготными носителями гена ФКУ. Ген фенилкетонурии встречается в среднем у
1-2 на 100 человек, но болезнь может возникнуть лишь в том случае, если и мать
и отец ребенка являются носителями этого гена, и ребенок унаследует его в
двойном наборе. Поэтому болезнь встречается значительно реже, чем распространен
ген. Больные ФКУ (обладатели двух патологических генов) могут иметь детей с
фенилкетонурией только при вступлении в брак с носителями таких же генов. При
вступлении в брак с лицами свободными от гена ФКУ, их дети не болеют этим
заболеванием [2].
Состояние здоровья у гетерозигот. На первый взгляд
гетерозиготные родители больных ФКУ, а также их братья и сестры абсолютно
здоровы. Пенроуз, однако, обнаружил в семье больных ФКУ у шести родственников
особый вид психического заболевания депрессивного типа, которое проявлялось
после 50 лет. Он предположил, что для гетерозигот характерен повышенный риск
заболевания психическими расстройствами. Однако в течение последних 50 лет
проблеме возможных аномалий и подверженности заболеваниям у гетерозигот по ФКУ
уделялось поразительно мало внимания, и немногие исследования, посвященные этой
проблеме, часто не адекватны с точки зрения эпидемиологии [8].
Особенности, выявляемые у гетерозигот по ФКУ,
подразделяются на 4 основных типа: отклонения по IQ и другим психологическим тестам; повышенный риск
психических заболеваний; отклонения от нормы в электроэнцефалограмме (ЭЭГ),
нарушение процессов размножения. Отметим лишь, что большинство гетерозигот
психически здоровы, однако для них высок риск заболеть шизофренией
определенного типа, развивающейся в позднем возрасте. Описано небольшое
снижение IQ (особенно в отношении устной
речи), довольно часто выявляются отклонения в ЭЭГ. Согласно некоторым данным,
для гетерозиготных по ФКУ женщин повышен риск спонтанных абортов и
мертворождений [2, 6].
Заслуживают внимания отдельные сообщения об аномально
высоком уровне фенилаланина крови у гетерозигот в стрессовых ситуациях, таких
как грипп с высокой температурой или беременность. Специалистами в области
экогенетики было выдвинуто интересное предположение, что аспартам,
подслащивающее вещество с высоким содержанием фенилаланина, может наносить вред
развивающимся эмбрионам у гетерозиготных женщин. Приведенные здесь данные,
касающиеся гетерозигот по ФКУ, пока нельзя считать доказанными. Обследование
нескольких сотен детей с небольшим умственным отставанием и отклонениями в
поведении показало наличие среди них индивидов с повышенным уровнем
фенилаланина в крови. Для проверки этих данных необходимы дополнительные
исследования. Если факты подтвердятся, можно будет с уверенностью сказать, что
уменьшенная активность фермента у гетерозигот делает их более подверженными
определенным стрессовым ситуациям: этот феномен необходимо изучать [4, 8].
Лечение
фенилкетонурии и прогноз
Если ничего не предпринимать, фенилкетонурия приводит
к весьма тяжелым последствиям - развивается олигофрения. К счастью, этот
трагический исход можно предотвратить, если поставить правильный диагноз при
рождении. Главным способом лечения является диетотерапия, ограничивающая
поступление в организм фенилаланина. Приступить к ней нужно немедленно после
установления диагноза. При ранней диагностике это гарантирует нормальное
нервно-психическое развитие ребенка.
Диетотерапия, как единственный эффективный метод
лечения ФКУ, должна применяться с первых месяцев жизни ребенка, тогда поражение
мозга не разовьется. Важно ограничить количество потребляемого фенилаланина
таким образом, чтобы обеспечить его поступление в организм в количествах,
необходимых и достаточных для роста и развития, но предотвратив его накопление
в жидкостях тела. Кроме диетотерапии необходим постоянный медицинский контроль
за умственным и физическим развитием ребенка.
Очень важно! Применение диетотерапии на позднем этапе
не вернет ребенку нормального интеллекта. Дети, у которых это заболевание не
диагностируют сразу при рождении, а выявляют по умственной отсталости, не могут
быть излечены. По достижении 12-14 лет такие дети могут переходить на
нормальное питание и никаких признаков отравления фенилаланином у них не
наблюдается. Однако женщина, которая в детстве переболела ФКУ, должна снова
перейти на диету и употреблять только продукты с пониженным содержанием
фенилаланина перед зачатием, и оставаться на этой диете во время беременности и
кормления грудью. Если она не сделает этого, то ее ребенок подвергается риску
замедленного физического и умственного развития, даже если его отец не является
носителем гена ФКУ [15].
Единственным лечением, способным предотвратить
развитие слабоумия или уменьшить его степень, является диета, исключающая
поступление в организм фенилаланина сверх того минимального количества, которое
необходимо для образования собственных белков организма и его роста. Поэтому
смысл диетического лечения сводится к резкому ограничению естественного белка с
пищей. Для такого ограничения приходится полностью исключить из питания ребенка
такие богатые белками продукты как мясо, колбасы, рыбу, бульоны, яйца, творог,
сыр, мучные изделия, каши из естественных круп, фасоль, орехи, шоколад. Меню
для детей составляется из фруктов, овощей, крахмальных изделий, жиров, со
строгим учетом содержания в них фенилаланина.
Назначают белковые гидролизаты (цимогран, лефанолак,
берлофен, гипофенат) или аминокислотные смеси, лишенные фенилаланина, которые
становятся главными продуктами питания, обеспечивающими потребность в белке:
"Лофенолак", "Фенилфри" (США), "Берлофен",
"Апонти", "Гипофенат" у детей до 4-5 лет и
"Нофелан" - у детей старше 5 лет. Белковые гидролизаты вводят с
фруктовыми и овощными соками, пюре, супами.
Лечение проводят под контролем содержания фенилаланина
в крови, добиваясь поддержания его уровня в пределах 0,03-0,04 г/л. Строгое
ограничение белков животного происхождения требуется на протяжении первых 2-3
лет жизни. Наиболее рационально отменять диетическое лечение в возрасте 7-8 лет
[16, 17, 18].
Лекарственная
терапия:
Препараты с промедиаторным действием:
Наком (комбинация карбиДОФА и левоДОФА) -
доза 100-375 мг/сутки в течение 3-4 недель, перерыв между курсами 1,5-2 месяца;
Лево-дофа - доза 10-15 мг/кг в сутки;
5-окситриптофан - доза 10 мг/кг сутки.
- Ноотропные препараты
- Витамины, препараты кальция, фосфора, железа
- Препараты, улучшающие мозговое кровообращение (ноотропы –
церебролизин, аминолон, энцефабол)
- Препараты, улучшающие тканевой обмен (АТФ, рибоксин)
При диеторезистентных формах
лечение включает назначение тетрагидробиоптерина - доза 10-20 мг/кг в сутки.
Дети с фенилкетонурией способны усваивать только небольшие количества
фенилаланина, представленные в таблице 1:
Таблица 1. Допустимое суточное поступление
фенилаланина.
|
Возраст
ребенка |
Допустимое суточное количество
фенилаланина (мг/кг массы тела) |
|
||
|
До 2
мес. |
60 |
|
||
|
2-3
мес. |
60-55 |
|
||
|
3-6
мес. |
55-45 |
||
|
6-12
мес. |
45-35 |
||
|
1-1,5
года |
35-30 |
||
|
1,5-3
года |
30-25 |
||
|
3-6
лет |
25-15 |
||
|
Старше
6 лет |
15-10 |
||
Возможно ли грудное вскармливание при фенилкетонурии?
Грудное
вскармливание при фенилкетонурии возможно и даже
приветствуется, однако только в том случае, если мать больного ребенка будет
придерживаться строгих правил кормления. В частности, необходимо строго
контролировать количество грудного молока, которое получает ребенок. Для этого
кормление проводится только сцеженным молоком, которое дается ребенку в строго
определенном количестве. Для покрытия энергетических потребностей ребенка к
разрешенному количеству грудного молока или обычной молочной смеси добавляют
необходимое количество специализированной смеси – гидролизата белка, которая не
содержит фенилаланина. По мере роста ребенка грудное молоко постепенно
полностью заменяют на специализированную смесь.
Расчет необходимого объема молока в зависимости от
возраста и веса ребенка
Общий объем питания (грудное молоко + специальная
смесь) должен соответствовать возрасту ребенка, больного фенилкетонурией.
Пример: Ребенок 2 месяца, вес 4200гр. Допустимое количество
фенилаланина которое ребенок может получить в сутки: 60*4=240 мг. Так как в 100
мл грудного молока содержится 56 мг фенилаланина, то ребенок может получить:
(240*100)/56=428 мл молока в сутки.
Для приготовления смеси с гидролизатом белка
используют сцеженное грудное молоко, растительное масло, кукурузный крахмал,
сахар, воду, гидролизат белка. В растительное масло добавляют кукурузный
крахмал, сцеженное грудное молоко, сахар – все перемешивают. Добавляют воду Ѕ
от общего количества жидкости необходимого ребенку (таблица 2), кипятят 2
минуты. В оставшейся воде разводят гидролизат белка, кипятят и после кипячения,
смешивают обе жидкости. Приготовленную смесь разливают по бутылочкам и хранят в
холодильнике. Перед употреблением смесь нагревают до температуры 37 градусов
[15, 16].
Таблица 2.
Суточная потребность в жидкости.
|
Вес
младенца,кг |
Потребление жидкости в день, мл |
|
до 3,5 |
525 |
|
4 |
600 |
|
4,5 |
675 |
|
5 |
750 |
|
5,5 |
825 |
|
6 |
900 |
|
6,5 |
975 |
|
7 |
1050 |
|
7,5 |
1125 |
|
8 |
1200 |
|
8,5 |
1275 |
|
9 |
1350 |
|
9,5 |
1425 |
|
более 10 |
1500 |
Примерный суточный рацион
ребенка, больного фенилкетонурией, 4 - 6 месяцев жизни с массой тела 5-6 кг:
Грудное молоко – 350 г
Масло растительное – 10 г
Масло сливочное – 8 г
Кукурузный крахмал – 20 г
Рыбий жир – 1 ч.л
Белковый гидролизат – 23 г
Яблочный сок – 50 г
Яблочное пюре – 30 г
Сахар –
20 г
Глюкоза –
20 г
Для питания детей с фенилкетонурией используются
специальные продукты – гидролизаты белка или смеси аминокислот, полностью или
частично лишенные фенилаланина. Эти лечебные смеси приближены по составу к
грудному молоку и содержат все необходимые ребенку пищевые компоненты.
Для больных детей первого
года жизни используют:
Афенилак
Аналог – ХР;
Лофеналак
Продукты для детей старше
1 года:
Тетрафен;
Фенил-фри;
Максамум – ХР
Максамаид - ХР
Смесь «Максамум – ХР» рекомендуют использовать также и
для детей старше 6 лет и питания беременных женщин, больных фенилкетонурией.
Смеси аминокислот и гидролизатов белка вводят в рацион
ребенка постепенно. Начальная доза может составлять 1/3 – 1/5 от суточного
количества препарата. За неделю количество лечебной смеси доводят до
необходимого объема. Детям первого года жизни гидролизат белка добавляют в
каждый прием пищи. Детям старше 1 года смесь дают 2 раза в день – утром и в
полдник, разбавляя смесь соками или чаем.
Правило введения прикорма в питание детей,
больных фенилкетонурией.
Рацион ребенка, больного фенилкетонурией, после года
значительно отличается от диеты здоровых детей. Основу стола должны составлять
овощи и фрукты, а также безбелковые продукты (безбелковые макароны, саго,
кукурузный или амилопектиновый крахмал, безбелковый хлеб и кондитерские
изделия, безбелковые крупы). Дефицит жиров должен быть восполнен за счет
сливочного и растительного масел, а у детей первого года жизни — за счет
рыбьего жира. Введение прикорма у детей, больных
фенилкетонурией начинается с 4 месячного возраста: рацион расширяют за счет
фруктовых и ягодных соков (грушевый, яблочный, сливовый).
Первый прикорм в виде твердой пищи вводят в 4,5 - 5
месяцев: овощное пюре или плодовоовощные консервы, но без добавления молока.
Второй прикорм в 5,5 месяцев: 10%-ную кашу из молотого
саго, безбелковой крупы или безмолочной каши промышленного производства на
основе кукурузной и рисовой муки, которые содержат не более 1,0 г белка в 100
мл готового к употреблению продукта.
С 6-7 месяцев в питание вводят муссы, кисели, которые
готовятся с использованием амилопектинового набухающего крахмала и фруктового
сока. Овощи, фрукты и молоко вводят в диету на основании подсчета содержащегося
в них фенилаланина (1 гр белка содержит приблизительно 50 мг фенилаланина).
Содержание
фенилаланина в продуктах (в граммах на 100г продукта):
Молоко женское — 0,056
Крупа рисовая — 0,313
Крупа манная — 0,399
Крупа гречневая — 0,395
Крупа овсяная — 0,363
Крупа пшенная — 0,48
Крупа перловая — 0,331
Горох —
0,763
Мука пшеничная — 0,322
Макаронные изделия — 0,488
Хлеб ржаной — 0,278
Хлеб пшеничный — 0,330
Печенье —
0,334
Картофель —
0,083
Морковь —
0,059
Капуста белокочанная — 0,073
Помидоры —
0,023
Апельсины —
0,04
Лимоны —
0,032
Сок яблочный — 0,021
Сок апельсиновый — 0,042
Сок лимонный — 0,036
Контроль лечения фенилкетонурии.
Диетическое лечение детей проводят под строгим
контролем содержания фенилаланина в крови. Оптимальный показатель фенилаланина
должен быть в пределах 3-6 мг%, при его значительных колебаниях проводят
коррекцию белка в рационе ребенка, больного фенилкетонурией. Уровень
фенилаланина определяют каждый месяц в течение первого года жизни ребенка. У
детей старше 1 года исследование уровня фенилаланина проводят один раз в 2-3 месяца.
Все дети, больные фенилкетонурией, должны находиться под наблюдением
психоневролога и педиатра [16, 17, 18, 19, 21].
Генотерапия. Из 3 основных шагов, требуемых для
генотерапии при фенилкетонурии, 2 выполнены: получена кДНК, обеспечивающая
экспрессию гидроксилазы фенилаланина человека, разработана
гидроксилазадефицитная животная модель. Однако векторы для эффективной передачи
гена in vivo
требуют дальнейшей разработки. Ретровирусные векторы, несмотря на то, что
достаточно эффективны in vitro, имеют низкую эффективность передачи in vivo.
Рекомбинантные аденовирусные векторы, хотя и полностью успешны на короткое
время, не сохраняются в организме больше нескольких недель из-за иммунного
ответа [20].
Профилактика
фенилкетонурии
Выявление гетерозиготных носителей. Большое значение
имеет специальное наблюдение за семьями риска, т.е. за такими семьями, где уже
имелись дети с фенилкетонурией. Новорожденные из этих семей должны быть
подвергнуты обязательному биохимическому исследованию и при показаниях к раннему
лечению.
Внедрение программ массового скрининга новорожденных
для раннего выявления ФКУ и своевременного назначения диетотерапии. Выявление и
лечение детей по программам массового скрининга также позволяет предупредить
развитие тяжелой психической инвалидности.
Пренатальная диагностика: Предложен ДНК-зонд для
пренатальной диагностики фенилкетонурии в семьях высокого риска [8, 10].
Список
литературы
1.
Бадалян Л.О., Таболин В.А., Вельтищев Ю.Е. Наследственные болезни у детей. - М.
Медицина, 1971.- 321 с.
2.
Е.М. Мастюкова, А.Г.
Московкина. Основы генетики. – М. ВЛАДОС,
2005. – 367 с.
3.
Harding C. O. et al. Advances and challenges in
phenylketonuria // J. Inherit. Metab.
Dis. 2010. V. 33. P. 645–648.
4.
Blau N. et al. Phenylketonuria // Lancet. 2010. № 376. P. 1417–1427.
5.
Троицкая
Л.А. Динамика психического развития детей с фенилкетонурией под воздействием
медико-психологической коррекции: Автореф. …канд. мед. наук. – М., 1993. – 16с.
6.
Первичная
профилактика психических, неврологических и психосоциальных расстройств. \ Под.
ред. А.Н. Моховикова – М., 2002.
7.
Lord B. et al. Implications of resolving the diagnosis
of PKU for parents and children // J. Pediatr. Psychol. 2008. V. 33. P. 855–866.
8. McKusick
V. Mendelian inheritance in man. – Baltimore – London: Johns Hopkins University
Press, 1992. – 1981p.
9.
Koch R. et al. Neuropathology of a 4-month-old infant
born to a woman with phenylketonuria // Dev. Med. Child. Neurol. 2008. V. 50. P. 230–233.
10. Brassier A,
Ottolenghi C, Boddaert N, Sonigo P, Attiй-Bitach T, Millischer-Bellaiche AE, Baujat G,
Cormier-Daire V, Valayannopoulos V, Seta N, Piraud M, Chadefaux-Vekemans B,
Vianey-Saban C, Froissart R, de Lonlay P. Prenatal symptoms and diagnosis of
inherited metabolic diseases. Arch Pediatr. 2012 Sep;19(9):959-69.
11.
Allen R.J., Brunberg J., Schwartz E. et al. MRI
characterization of cerebral dysgenesis in maternal PKU// Acta Paediatr. Int. J. Paediatr. – 1994. –Vol. 83. –
Suppl. – P.83-85.
12. Levy H.L.,
Waisbren S.E., Lobbregt D. et al. Maternal mind hyperphnylalaninemia: an
international survey of offspring outcome// Lancet. – 1994. - Vol. 344. –
P.1589-1594.
13.
Waisbren S.E., Zaff J. Personality disorder in young
women with treated phenylketonuria//J. Inher. Merab. Dis. – 1994. – Vol. 17. – P. 584-592.
14. Waisbren S.E.,
Levy H.L. Effects of untreated maternal hyperphnylalaninemia on the fetus:
further study of families identified by routine cord blood screening//J.
Paediatr. – 1990. – Vol. 117. – P. 926-929.
15.
Лечебное
питание при наследственных нарушениях обмена// Клиническая диетология детского
возраста / Под ред. Боровик Т. Э., Ладодо К. С. М.: «МИА». 2008. С. 330–383.
16.
Singh R. H. et al. BH4 therapy impacts the nutrition
status and intake in children with phenylketonuria: 2-year follow-up // J.
Inherit. Metab. Dis. 2010. V.
33. P. 689–695.
17.
Webster D. et al. Tyrosine supplementation for
phenylketonuria // Cochrane Database Syst. Rev. 2010. V. 8: CD001507.
18.
Van Spronsen F. J. et al. Large neutral amino acids in
the treatment of PKU: from theory to practice // J. Inherit. Metab. Dis. 2010. V. 33. P. 671–676.
19.
Harding C. O. New era in treatment for
phenylketonuria: pharmacologic therapy with sapropterin dihydrochloride //
Biologics. 2010. V. 9. P. 231–236.
20. Eisensmith R. C.
and Woo S. L. C. Somatic Gene Therapy for phenylketonuria and other hepatic
deficiencies. Journal of Inherited Metabolic Diseases. 2005. №19(4), р.412-23.
21. Mütze U,
Beblo S, Kortz L, Matthies C, Koletzko B, Bruegel M, Rohde C, Thiery J, Kiess
W, Ceglarek U. Metabolomics of dietary Fatty Acid restriction in patients with
phenylketonuria. PLoS One. 2012;7(8).