Воробьева Д.В., Цыщук И.Е., Осипов
С.Н.
Федеральное
государственное бюджетное учреждение науки
Институт
элементоорганических соединений
им.
А.Н.Несмеянова Российской академии наук, Россия
Трифторметилсодержащие стирольные
лиганды для создания эффективных катализаторов метатезиса олефинов
В течение последнего десятилетия метатезис олефинов,
как важный метод образования новых углерод-углеродных связей, стал одной из
самых динамично развивающихся областей органической химии. Тем не менее, в ряде
превращений, например таких как, кросс-метатезис, метатезис полимеризации с
раскрытием цикла, а также метатезис разветвленных олефинов, до сих пор остается
весьма актуальной проблема поиска наиболее эффективного и селективного
катализатора. Ее успешное решение может привести к созданию новых
стереорегулярных полимеров с уникальными свойствами, промышленно важных
процессов утилизации побочных продуктов крекинга нефти, а также альтернативных
источников энергии на основе биоресурсов1. Поэтому разработка
простых методов синтеза новых недорогих катализаторов на основе рутения с
улучшенными характеристиками, особенно в наиболее проблемных типах метатезиса,
является весьма актуальной областью современной химической науки.
С другой стороны, в последнее время бурное развитие
получила химия фторорганических соединений2. В настоящее время
фторсодержащие вещества уже нашли широкое применение практически во всех
областях народного хозяйства, включая медицину и производство новых материалов.
Это связано, прежде всего с тем, что введение атомов фтора или фторалкильных
групп в молекулу существенно меняет ее физико-химические свойства. Особенно это
касается трифторметилированных соединений3, так как CF3-группа
является одной из самых липофильных групп в органической химии, обладает
высокой электроотрицательностью и большим стерическим объемом (близким к
изопропильной группе). Кроме того, фтор способен образовывать достаточно
прочные, как водородные связи с окружающими функциональными группами, так и
координационные связи с различными металлами, включая рутений. В связи с этим
разработка эффективной стратегии синтеза новых трифторметилзамещенных
производных стирола с окси- или аминофункцией в орто-положении, которые
способны выступать в качестве лигандов для получения новых карбеновых комплексов
рутения является весьма актуальным. С этой целью нами были изучены реакции
внедрения CF3-карбена, каталитически генерируемого in situ из CF3-содержащего
2-диазокарбоксилата, в OH- и NH-связи соответствующих ароматических соединений.
Из литературы известно,
что внедрение металлокарбеноидов в ОН-связи органических молекул происходит,
как правило, в более мягких условиях, чем реакции циклопропанирования4.
Поэтому, наши исследования были начаты с реакции трифторметилсодержащего
диазокарбоксилата с 2-гидроксистиролом, в расчёте на преимущественное
образование желаемого продукта внедрения соответствующего CF3-содержащего карбена по фенольной ОН-группе 2, а не по двойной связи 3
(Схема 1).
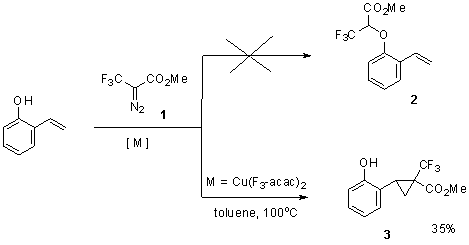
Схема 1
В результате было обнаружено, что реакция
эквимольных количеств 2-гидроксистирола и трифтордиазокарбоксилата 1 осуществляется в толуоле при 110оС
в присутствии медного катализатора Cu(F3-acac)2
в количестве 5 мольн.% и завершается за 15-20 минут, приводя к смеси продуктов,
из которой удалось выделить и полностью охарактеризовать лишь продукт
присоединения карбена по двойной
связи 3 с выходом 35% после очистки
на колонке с силикагелем.
Следующая
попытка синтеза целевого стирола 2
заключалась в возможной реализации подхода, представленного на схеме 2. Метод
включал первоначальное внедрение CF3-карбена в ОН-связь фенола 4, ортоформилирование 5 в
трифторуксусной кислоте с использованием уротропина в качестве источника
формильной группы и последующую реакцию Виттига. Несмотря на то, что первая
стадия завершалась селективным образованием продукта внедрения 5 с хорошим выходом, последующие стадии
завершались образованием продуктов 6 и
7 с низкими выходами даже после
попыток их оптимизации.
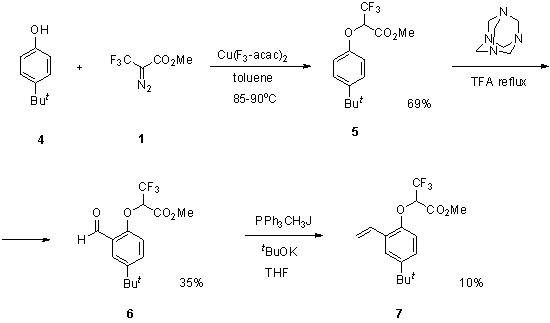
Схема 2
Поэтому
нами был апробирован еще один синтетический подход, основанный на использовании
орто-аллилфенола 8. Выбор аллилфенола в качестве исходного соединения обусловлен
наличием менее активной к циклопропанированию аллильной двойной связи по
сравнению с двойной связью стирола, а также возможностью изомеризации
аллилбензольного производного в соответствующий β-метилстирол в
присутствии комплексов металлов переходной группы. Внедрение трифторсодержащего
карбена по ОН-связи осуществляли, используя описанные выше условия. Оказалось,
что в отличие от 2-гидроксистирола реакция приводит к образованию желаемого
продукта внедрения 9, выход которого
после стадии очистки составил лишь 32%. Тем не менее, соединение 10 было наработано в достаточном
количестве для проведения следующей стадии изомеризации (Схема 3).
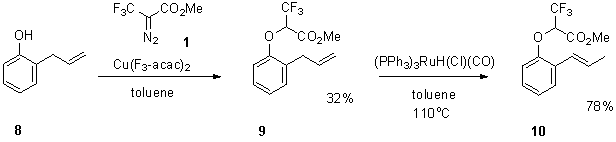
Схема
3
Каталитическую
изомеризацию 9 в 10 осуществляли в присутствии
гидридного рутениевого комплекса (PPh3)3RuH(Cl)(CO) при
кипячении в толуоле в течение 10 часов, приводящую к желаемому
трифторметилсодержащему стиролу 10 с
78% выходом после очистки. Следует отметить, что несмотря на наличие метильной
группы в β-положении, стирол 10
можно рассматривать в качестве функционального эквивалента стирола 2 (схема 1) в лигандном обмене при
конструировании новых карбеновых комплексов рутения, так как реакция реализующая
по механизму кросс-метатезиса с выделением пропилена (в случае 10) или этилена (в случае 2), приводит к одному и тому же
продукту5.
Также
мы обнаружили, что стирол 10 может
быть получен прямой функционализацией коммерчески доступного 2-пропенилфенола.
Оказалось, что в отличие от 2-винилфенола (см. выше), в данном случае атака CF3-карбена осуществляется
исключительно по гидроксильной группе, приводя к целевому продукту с хорошим
выходом. Вероятно, наличие метильной группы в исходном стироле препятствует
образованию циклопропанового производного (Схема 4).
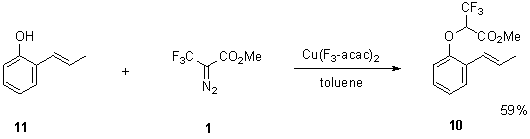
Схема
4
Также в качестве лигандов хелатного типа для
направленной модификации рутениевых катализаторов метатезиса олефинов могут
выступать и производные аминостирола6. В связи с этим, нами были
предложены две альтернативные синтетические последовательности получения соответствующего
стирола 14 из коммерчески доступного
2-броманилина (Схема 5). В первом случае обработкой трибутилвиниловом 2-броманилина
нами был получен аминостирол 12 с
40%-ным выходом, который далее включали в реакцию с диазокарбоксилатом 1. При этом было обнаружено, что в
отличие от 2-гидроксистирола (Схема 2) преимущественно образуется желаемый
продукт внедрения CF3-карбена по NH-группе
аминостирола 14 с 46% выходом.
Второй подход к аминостиролу 14 включал
NH-внедрение CF3-карбена, образующегося
при каталитическом разложении соответствующего диазосоединения, в 2-броманилин,
давая 13 с 75%-ным выходом. Последующая
Pd-катализируемая реакция 13
с трибутилвинилоловом привела к образованию целевого продукта 14 с хорошим выходом.
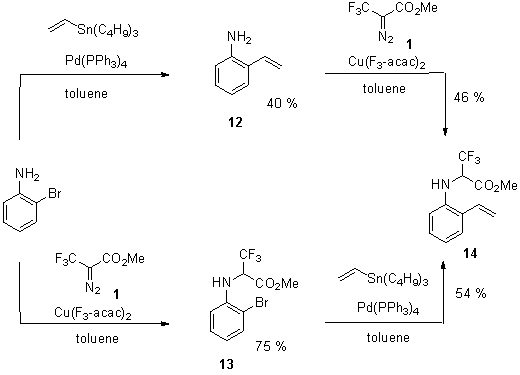
Схема 5
Полученный аминостирол 14, модифицированный CF3-группой, далее включили
в лигандо-обменную реакцию, представленную на схеме 6, в присутствии хлорида
меди и инденилиденового комплекса 15
второго поколения, который выступает в качестве источника рутения. После нагревания
в течение часа при 40 ºС нами был выделен комплекс 16 с 20%-ным выходом после очистки на колонке с силикагелем.
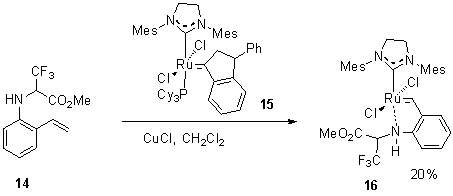
Схема 6
В настоящий момент нами осуществляется
оптимизация синтеза комплекса 16, а также исследования его
каталитической активности.
В результате проведенного исследования нами
разработаны удобные методы синтеза новых трифторметилсодержащих гидрокси- и
аминостиролов, перспективных лигандов хелатного типа для направленной
модификации рутениевых катализаторов метатезиса олефинов. Метод основан на реакции
селективного внедрения CF3-карбена, каталитически
генерируемого из метилтрифтордиазопропионата в ОН- и NH-связи коммерчески
доступных 2-пропенилфенола и 2-броманилина.
Работа
выполнена при финансовой поддержке Российского Фонда Фундаментальных
Исследований (проекты №№ 12-03-31103-мол-а, 12-03-00557, 12-03-93111).
Литература:
1. а) Endo K.,
Grubbs R.H., J. Am. Chem. Soc. 2011, 133, 8525; b)
Rouhi A.M., Chem. Eng. News 2002, 80, 34; c) Gottumukkala
A.L., Madduri A.V.R., Minnaard A.J., ChemCatChem.
2012, 4, 462; d) Bruneau C., Fischmeister C.,
Miao X., Malacea R., Dixneuf P.H., Eur.
J. Lipid Sci. Technol. 2010, 112, 3; e) Miao X., Fischmeister C., Bruneau C., Dixneuf P.H.,
ChemSusChem. 2009, 2, 542.
2. Kirsch
P., Modern Fluoroorganic Chemistry;
Wiley-VCH: Weinheim, Germany, 2004;
b) Purser S., Moore P.R., Swallow S., Gouverneur V., Chem. Soc. Rev. 2008, 37, 320; c) Kirk K.L., J. Fluorine Chem. 2006, 127, 1013, d)
Shimizu M., Hiyama T., Angew. Chem., Int.
Ed. 2005, 44, 214, e) Ma J.-A., Cahard D., Chem. Rev. 2004, 104, 6119, f) O’Hagan D., Chem. Soc. Rev., 2008, 37, 308, g) Nie J., Guo H.-C., Cahard D.,
Ma J.-A., Chem. Rev., 2011, 111, 455.
3. a)
Banks R.E., Tatlow J.C., Smart B.E., In
Organofluorine Chemistry: Principles and Commercial Applications; Plenum:
New York, NY, 1994, b) Uneyama K., Organofluorine Chemistry; Blackwell: Oxford, UK, 2006, c) Jagodzinska M., Huguenot F.,
Candiani G., Zanda M., ChemMedChem., 2009, 4(1), 49.
4. Ye T., McKervey M.A., Chem. Rev., 1994, 94, 1091.
5. Bieniek M., Samojłowicz C., Sashuk V., Bujok R.,
Śledź P., Lugan N., Lavigne G., Arlt D., and K. Grela, Organometallics,
2011, 4144-4158.
6. K. Żukowska, A. Szadkowska, A.E. Pazio, K.
Woźniak, and K. Grela, Organometallics,
2013, 462-469.