Садуакасова
А.Т., д.т.н. Самойлов В.И.
Восточно-Казахстанский
государственный технический университет
им. Д. Серикбаева, Казахстан
Физико-химические
основы ионообменных процессов химико-металлургических производств
На ранних стадиях развития
урановой технологии единственным методом выделения урана из растворов после выщелачивания
его из руд был осадительный метод, суть которого заключается в осаждении урана
в виде труднораствори-мых соединений [1]. Основные недостатки, свойственные для
этого метода – большой расход химического реагента-осадителя и сравнительно
низкое содержание урана в осадке. Позднее в урановой промышленности были
внедрены более прогрессивные методы – сорбционный (ионообменный) и
экстракционный [2, 3].
Сорбционный метод
извлечения урана из пульп и растворов начал внедряться в промышленность с 1952
г. Сорбция – это поглощение растворённых веществ твёрдыми материалами (сорбентами)
[1, 3, 4]. Сущность сорбционного метода состоит в следующем. Сорбент
контактируется с раствором или пульпой, содержащими извлекаемый металл. При
этом происходит сорбция металла на сорбенте. Раствор или пульпа, после
извлечения металла (рафинат), сбрасывается (рис. 1). Сорбент, насыщенный
извлекаемым металлом, промывается водой для удаления трудносорбируемых ионов и твёрдых
механических примесей. После промывки проводится десорбция металла с сорбента
(рис. 1). Десорбцию (элюирование) проводят раствором реагента, содержащим
катион или анион, который входит в состав исходного сорбента. При десорбции
происходит вымывание извлекаемого металла и регенерация сорбента до первоначального
его состояния. После десорбции сорбент снова возвращается на сорбцию металла
(рис. 1).
Раствор
Исходный
химического раствор раствор Н2О реагента
![]()
![]()
![]()
![]()
![]()
![]()
Промывка Десорбция Сорбция
Наыщенный
Сорбент сорбент,
Насыщенный
![]()
![]()
![]()
![]() примеси сорбент
примеси сорбент
![]()
![]()
![]()
![]()
Рафинат Н2О,
примеси Раствор,
содержащий
Сорбент извлекаемый
![]() металл
металл
Рисунок 1 – Общая
схема сорбционного концентрирования металлов [1]
Лежащие в основе сорбции ионообменные процессы занимают важное
место в атомной технологии. Кроме указанного применения для извлечения урана
из растворов (пульп), получаемых при выщелачивании урановых руд, ионообменные
процессы используются для полного обессоливания воды на АЭС, для улавливания
радиоактивных изотопов из сточных вод атомных предприятий [3,
5].
Ионообменные процессы извлечения и очистки
металлов от примесей основаны на способности ряда твёрдых веществ (ионитов)
обмениваться ионами с растворами электролитов [2].
Иониты
– природные или искусственные неорганические или высокомолекулярные органические
соединения, практически нерастворимые в воде и водных растворах кислот, щелочей
и солей, содержащие ионогенные группы, способные к эквивалентному обмену
ионами, растворёнными в электролите. В зависимости от вида ионогенной активной
группы иониты подразделяют на классы – катиониты и аниониты [2, 6]. Существуют
также биполярные иониты (амфолиты), имеющие и катионообменные и анионообменные
группы [7].
В
настоящее время в промышленности в качестве ионитов используют такие
соединения как цеолиты, сульфоугли, ионообменные смолы. Смолы получили
наибольшее распространение в производстве цветных и редких металлов [2].
Макромолекула ионообменной
смолы состоит из гибких переплетённых нитей полимерных молекул, углеводородные
цепи которых имеют поперечные связи, образующие сетчатую структуру, так
называемую матрицу. Матрица содержит неподвижные заряженные группы, природу и
количество которых можно регулировать при синтезе смол. Заряд фиксированных
ионов уравновешивается подвижными ионами противоположного знака
(противоионами), способными вступать в ионообменные реакции с ионами,
находящимися в электролите (рис. 2).
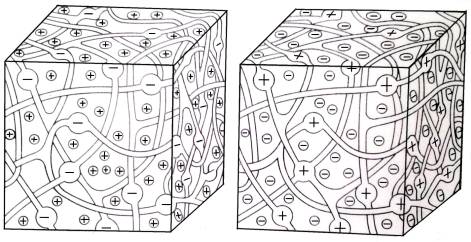
а б
Рисунок 2 –
Трёхмерная модель структуры катионита (а) и анионита (б) [2]
Активными группами в
катионитах являются: сульфогруппа (–SO3H),
карбоксильная группа (–СООН), гидроксильная группа (–ОН) и другие, более
сложные соединения. Аниониты в качестве активных групп содержат аминогруппы
разной степени замещения (–NH2), (=NH),
(≡N), четвертичные аммониевые основания (![]() N+) и др.
N+) и др.
В зависимости от
назначения в промышленности применяют смолы с частицами размером от 0,3 до 2,0
мм, суммарная поверхность которых изменяется от 0,2 до 300 м2/г.
Важнейшей
характеристикой ионита является так называемая обменная ёмкость ионита. Она
выражает количество задержанных ионитом ионов в принятых условиях. Различают
три вида обменной ёмкости:
1) статическая ёмкость
(СОЕ) представляет собой количество ионов, сорбированных единицей смолы,
находящейся в равновесии с электролитом;
2) динамическая ёмкость
(ДОЕ) выражает количество ионов, сорбированных единицей смолы при
контактировании электролита со смолой в динамических условиях (фильтрации) до
появления (проскока) сорбируемого иона в растворе, прошедшего сквозь слой
смолы;
3) полная динамическая
ёмкость (ПДОЕ) выражает количество сорбированных ионов в динамических условиях
до полного насыщения слоя смолы.
Принято считать, что смола достигает ПДОЕ, если
концентрация подлежащего поглощению иона в выходящем растворе равняется
концентрации этого иона в исходном растворе. На практике ёмкость ионита
выражают в единицах массы на единицу объёма набухшей смолы (г/л, кг/м3).
Обменная ёмкость смол в
значительной мере зависит от рН среды [2, 7, 8]. Эта зависимость положена в
основу классификации смол, причём по характеру зависимости СОЕ от рН среды все
смолы подразделяются на четыре класса (рис. 3).
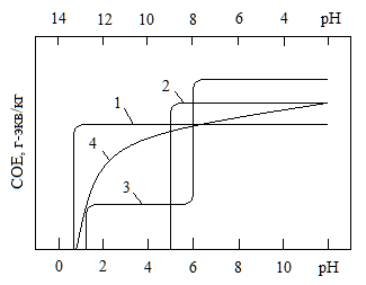
1 – иониты I
класса; 2 – иониты II класса; 3 – иониты III
класса; 4 – иониты IV
класса
Рисунок 3 – Зависимость
СОЕ смол от рН среды [2]
К I
классу относятся иониты, проявляющие свойства сильных кислот или оснований. Их
обменная ёмкость остаётся постоянной в широком интервале значений рН. Такие
свойства катиониту придаёт группа –SO3H, а
аниониту группа ![]() N+.
N+.
Ко II
классу относятся иониты, проявляющие свойства слабых кислот или слабых
оснований. Их обменная ёмкость достигает максимума только при определённом
значении рН раствора, высоким для катионитов и низком для анионитов. Такие
свойства катионитов связаны с наличием
в них активных групп –СООН и –ОН, которые могут обменивать ионы водорода на
другие катионы только в нейтральной или щелочной среде. Для анионитов с активными
группами –СООН и –ОН характерен обмен ионов ОН– на другие анионы
только в нейтральной или кислой среде.
К двум классам (III
и IV) относятся иониты смешанного типа. Иониты III
класса
ведут себя подобно смесям многих кислот или оснований различной силы. Обменная
ёмкость их меняется в широком интервале значений рН.
Ионообменное равновесие. При контактировании
ионита с раствором электролита на границе раздела фаз устанавливается разность
электрических потенциалов, вызванная перераспределением ионов между твёрдой и
жидкой фазами (потенциал Доннана) [2, 7]:
|
|
(1) |
где ![]() , μi – химический потенциал i-того
иона в жидкой и твёрдой фазах; φ – электрический потенциал;
Zi – электрический заряд i-того
иона; F – число Фарадея.
, μi – химический потенциал i-того
иона в жидкой и твёрдой фазах; φ – электрический потенциал;
Zi – электрический заряд i-того
иона; F – число Фарадея.
Данная разность
потенциалов противодействует стремлению ионов сравнять свои концентрации в
ионите и растворе вследствие диффузионного переноса вещества. Таким образом,
при ионообменных процессах на ионы действуют с одной стороны градиент
концентрации, вызывающий диффузию. И с другой стороны – электростатические
силы, противодействующие диффузии ионов из смолы. Вследствие этого на границе
раздела ионит–раствор устанавливается равновесие, при котором для каждого вида
ионов воздействие кулоновских сил уравновешивается диффузионным потоком.
В случаях, когда
химический потенциал ионов зависит от состава раствора, а поверхность зерна
ионита можно представить в виде полупроницаемой перегородки, которая
пропускает сквозь себя молекулы растворителя и задерживает на себе растворённые
ионы, величина разности электростатического потенциала может быть представлена
зависимостью:
![]() – φ = RT/(ZiF) (ln ai/
– φ = RT/(ZiF) (ln ai/![]() – πVi), (2)
– πVi), (2)
где ai, ai – активности i-того
иона в растворе и фазе ионита; π
– давление набухания; Vi – парциальный молярный
объём i-того иона.
Используя приведённое выше
уравнение для раствора, содержащего два противоиона А и В, где В – микропримесь,
получают выражение для оценки эффективности разделения ионов и противоионов А и
В, т. е. выражение для расчёта коэффициента разделения:
ln α = ln ![]() ] +
] +
+ π/(RT![]() ) ∙ (
) ∙ (![]() ), (3)
), (3)
где α – коэффициент разделения;
m – молярность
ионов; γ – коэффициент активности
ионов.
Влияние на коэффициент разделения
отдельных составляющих следующее.
Из уравнения следует, что с
разбавлением исходного раствора коэффициент селективности возрастает, т. к.
уменьшается величина mA. С увеличением значения ![]() , т. е. числа фиксированных ионов в
матрице смолы, коэффициент разделения также возрастает.
, т. е. числа фиксированных ионов в
матрице смолы, коэффициент разделения также возрастает.
С уменьшением коэффициента активности В-иона в фазе ионита растёт сорбция
микропримеси. К тем же результатам приводит и увеличение ![]() . Снижение активности
. Снижение активности ![]() может быть достигнуто за счёт ассоциации В-ионов с фиксированными ионами или образования ионных пар между
фиксированными ионами и ионами микропримеси. Такого рода взаимодействия
сопровождаются локализацией ионов примеси около фиксированных ионов и способствует
снижению коэффициента активности
может быть достигнуто за счёт ассоциации В-ионов с фиксированными ионами или образования ионных пар между
фиксированными ионами и ионами микропримеси. Такого рода взаимодействия
сопровождаются локализацией ионов примеси около фиксированных ионов и способствует
снижению коэффициента активности ![]() . Поэтому при разработке процессов по
удалению микропримесей из растворов необходимо выбирать такие смолы, которые
содержат активные группы, способные образовать с ионами микропримеси прочные
комплексные или малорастворимые соединения.
. Поэтому при разработке процессов по
удалению микропримесей из растворов необходимо выбирать такие смолы, которые
содержат активные группы, способные образовать с ионами микропримеси прочные
комплексные или малорастворимые соединения.
Образование
прочных внутрикомплексных соединений между микропримесями и активными группами
смол может привести к сорбции примеси катионов и нейтральных и слабокислых сред
даже анионитами.
Степень
очистки растворов от микропримесей в значительной степени зависит от
регенерации ионита, т. е. от остаточной концентрации удаляемых микропримесей в
фазе ионита. С понижением начальной концентрации микропримесей в очищаемом
растворе для сохранения эффективности очистки необходимо соответственно
уменьшать и остаточную концентрацию микропримесей в ионите. Таким образом,
очистка растворов от микропримесей зависит от совершенства не только от
процесса их извлечения на смолы, но и обратного процесса – десорбции их со
смолы.
Из уравнения также следует, что с увеличением заряда
примеси ![]() и уменьшением заряда основного
вещества
и уменьшением заряда основного
вещества ![]() возрастает коэффициент разделения.
возрастает коэффициент разделения.
Кинетика ионного обмена. Анализ работ по изучению кинетики ионного обмена [2, 6-8]
показывает, что этот процесс протекает с измеримой скоростью. Если сферическую
частицу ионита погрузить в поток жидкости (рис. 4), то скорость потока
жидкости будет уменьшаться по мере приближения потока к границе раздела
жидкость–твёрдое и вблизи этого раздела станет равной нулю, т. е. вокруг зерна
ионита образуется неподвижный (пограничный) слой жидкости.
Если один из двух противоионов более подвижен, то его поток
в начальный момент должен быть большим, но благодаря этому возникает кратковременный
пространственный заряд, который тормозит более быстрый ион и ускоряет более медленный
[3, 7, 9]. Таким образом, происходит выравнивание скоростей движения
более подвижного и менее подвижного ионов. Процесс ионного обмена состоит из
взаимодиффузии ионов А и В как в зерне ионита, так и в диффузионной пленке. Если
самой медленной стадией является процесс взаимодиффузии в плёнке, то считается,
что процесс управляется плёночной или внешнедиффузионной кинетикой. Если же
самой медленной
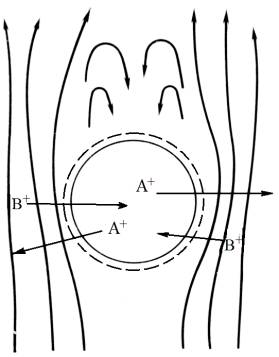
Рисунок 4 – Обтекание частицы ионита
потоком раствора [2]
стадией
является взаимодиффузия в зерне ионита,
то процесс управляется гелевой или внутридиффузионной кинетикой. Лимитирующую стадию процесса
можно предсказать теоретически, используя неравенства [2]:
при гелевой диффузии
[(![]() δ)/(CDr0)] (5 + 2αA/B)
δ)/(CDr0)] (5 + 2αA/B) ![]() 1, (4)
1, (4)
при плёночной диффузии
[(![]() δ)/(CDr0)] (5 + 2αA/B)
δ)/(CDr0)] (5 + 2αA/B) ![]() 1, (5)
1, (5)
где D –
коэффициент диффузии; αА/В – коэффициент разделения; С –
концентрация противоиона; r0 –
размер зерна ионита; δ – толщина диффузионного слоя.
При чисто плёночной
кинетике ионного обмена, модель которой представлена на рис. 5, можно считать,
что градиент концентрации обменивающихся ионов в объёме зерна отсутствует, т.
е. СА = САср ≈ С’А. Если
принять, что эффективная толщина диффузионного слоя значительно меньше радиуса зерна
и считать поверхность последнего плоской, а также что концентрация ионов на
внешней границе плёнки поддерживается постоянной, то для кине-
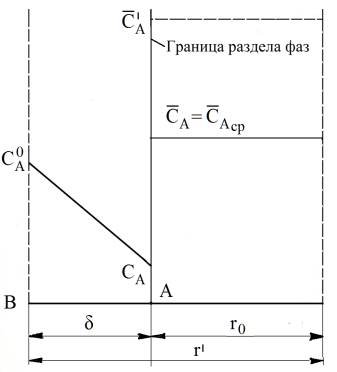
r – радиус частицы ионита; ![]() – концентрация иона А
в ядре потока;
– концентрация иона А
в ядре потока; ![]() – концентрация иона А на границе жидкость –
ионит;
– концентрация иона А на границе жидкость –
ионит; ![]() – концентрация иона
А в ионите, равновесная с
– концентрация иона
А в ионите, равновесная с ![]() ;
; ![]() – средняя
концентрация иона А в ионите
– средняя
концентрация иона А в ионите
Рисунок 5 – Изменение
концентрации ионов при переходе из жидкой фазы в зерно при плёночной диффузии [2]
тики сорбции можно получить уравнение в виде [2]:
![]() =
=![]() (1–
(1– ![]() ) =
) = ![]() (1–
(1– ![]() τ/α), (6)
τ/α), (6)
где
К1 – константа скорости (К1 – 3D/δαr0); α
– коэффициент распределения; β1 – коэффициент скорости для
плёночной области (β1 = К1α).
Из этого уравнения
видно, что процесс будет ускоряться при увеличении концентрации ионов в
исходном растворе, росте подвижности обменивающихся ионов, повышении
интенсивности перемешивания раствора (приводящее к уменьшению толщины
диффузионного слоя), уменьшении крупности зёрен ионита. При прочих равных
условиях относительная скорость обмена меньше у ионитов, имеющих большую
ёмкость и проявляющих более высокую избирательность по отношению к поглощаемым
ионам, характеризуемым соответственно большими значениями ![]() и К1.
и К1.
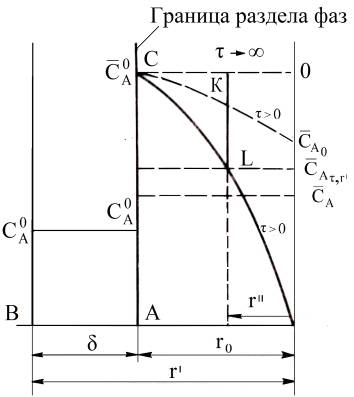
При чисто гелевой
кинетике градиенты концентраций в плёнке можно считать близкими к нулю. Изменение
концентрации по радиусу зерна ионита при неизменной концентрации внешнего
раствора на границе раздела фаз (рис. 6) рассчитывают по уравнению:
![]() (��2
(��2![]() )/(��r2); (7)
)/(��r2); (7)
для шарообразных частиц его можно описать в следующем
виде:
F = 1 – 6/π2![]() exp(–
exp(–![]() ), (8)
), (8)
где F = qτ/q∞ –
отношение сорбированного количества вещества за время τ (qτ) к
равновесному количеству сорбированного вещества; D – коэффициент внутренней диффузии; n – числа натурального ряда;
τ –
время; r – радиус частицы.
r – радиус частицы ионита; ![]() – концентрация иона
А в ядре потока;
– концентрация иона
А в ядре потока; ![]() – концентрация иона А на границе жидкость –
ионит;
– концентрация иона А на границе жидкость –
ионит; ![]() – концентрация иона
А в ионите, равновесная с
– концентрация иона
А в ионите, равновесная с ![]() ;
; ![]() – средняя
концентрация иона А в ионите
– средняя
концентрация иона А в ионите
Рисунок
6 – Изменение концентрации иона А
в зерне ионита при гелевой диффузии [2]
Зная D и r0 для заданного времени τ,
находят значение безразмерного параметра х: х = Dn2 τ/
r02 (в литературе по ионному обмену его обычно обозначают
через Bt) и и далее степень обмена, равную F(x).
Из рассмотренных
уравнений [2] следует, что относительная скорость ионного обмена, протекающего
в режиме гелевой кинетики, пропорциональна коэффициенту внутренней диффузии и
обратно пропорциональна квадрату радиуса зерна.
Литература:
1. Маслов А.А. Технология урана и плутония: учебное пособие / А.А. Маслов, Г.В. Каляцкая, Г.Н. Амелина, А.Ю. Водянкин,
Н.Б. Егоров – Томск: Издательство Томского политехнического университета, 2007.
– 97 с.
2. Матвеев Ю.Н., Стрижко
В.С. Технология металлургического производства цветных металлов (теория и
практика): Учебник для вузов. – М.: Металлургия, 1986. – 368 с.
3. Смирнов А.Л., Волкович В.А. Переработка облучённого
ядерного топлива. Конспект
лекций. – Екатеринбург: ГОУ ВПО УГТУ-УПИ, 2008. – 317 с.
4. Химическая энциклопедия (в 5 т.): т. 1: Под
ред. Кнунянц И.Л. – М.: Сов. Энцикл., 1988. – 623 с.
5. Сальникова Е.В., Мурсалимова М.Л., Стряпков
А.В. Методы концентрирования и разделения микроэлементов: учебное пособие. –
Оренбург: ГОУ ОГУ, 2005. – 157 с.
6.
Вдовенко В.М. Современная радиохимия. – М.: Атомиздат, 1969. – 544 с.
7.
Баймаков Ю.В. Металлургия редких металлов. Конспект лекций. – Л.: ЛПТИ им. М.И. Калинина. – 1969. – 165 с.
8.
Тураев
Н.С., Жерин И.И. Химия и технология урана: Учебное пособие для вузов –
М.:ЦНИИАТОМИНФОРМ, 2005. – 407 с.
9. Громов Б.В. Введение в химическую технологию урана.
Учебник для вузов. – М.: Атомиздат, 1978. – 336 с.